All published articles of this journal are available on ScienceDirect.

Evaluation of Madurahydroxylactone as a Slow Release Antibacterial Implant Coating
Authors Info & Affiliations
Abstract
Madurahydroxylactone (MHL), a secondary metabolite with antibacterial activity was evaluated for its suitability to generate controlled drug release coatings on medical implant materials. A smooth and firmly attached layer could be produced from a precursor solution on various metallic implant materials. In physiological salt solutions these coatings dissolved within a time period up to one week. A combination of MHL with a broad spectrum fluoroquinolone antibiotic was used to create a coating that was active against all bacterial strains tested. The time period during which the coating remained active against Pseudomonas aeruginosa was investigated. The results indicated a delayed drug release from single layer coatings in the course of seven days. MHL was biocompatible in cell culture assays and could after a delay even serve as a cell adhesion substrate for human or murine cells. The findings indicate a potential for MHL for the generation of delayed release antimicrobial implant coatings.
INTRODUCTION
Medical implant infections by a variety of microbes such as S. aureus, S. epidermidis, P. aeruginosa, coliforms and others still present a significant and a difficult to treat clinical problem [1-3]. Immediately after implantation various proteins present in blood serum or in other extracellular liquids readily adhere to the implant surfaces. It is thought that during this early phase after implantation before a layer of cells can form, a protective barrier to microbial invasion bacteria can most readily colonize the implant surfaces by adhering to the material surface or to extracellular proteins [4, 5]. When microbes can establish themselves on an implant surface they communicate with each other. In a process called quorum sensing secreted substances coordinate the bacterial behavior depending on the cell density. At higher cell densities a biofilm forms consisting of bacteria within a matrix of secreted polysaccharides. Microbes in biofilms show an increased resistance to conventional antibiotic treatments and also to the immune defense, whereby the quorum sensing substances secreted by the bacteria may act to down-regulate the immune response [6, 7]. Frequently the only effective treatment is the removal of the implant followed by antibiotic treatment to eliminate the infection [1, 8, 9]. This prolongs the suffering of the patient and increases the overall treatment costs. Antimicrobial coatings are a promising strategy to prevent biofilm formation on implants [10]. Such coatings allow high local antibiotic concentrations that are essential for the eradication of refractory microbial biofilm communities, while dilution effects are keeping systemic antibiotic levels below critical toxic values. However, even though the time period shortly after implantation is considered most critical, bacterial infections can occur well after wound healing is completed. Delayed or late implant infections are thought to arise by primary infections at other sites like the oral cavity and by diverting and weakening the immune defense or by direct dissemination of the infectious microbes by the blood circulation to the implantation site [11-13]. To prevent infections after surgery, basic antibiotic implant coatings are not suitable since these are rapidly released in a single burst. Consequently, numerous strategies have been devised to prolong the efficacious period by controlling the release of bioactive substances [14, 15]. These include (i) passive coatings whereby anti-adhesive or antimicrobial molecules are covalently bonded to the implant surface or by using meso-porous surfaces loaded with drugs (ii) metallic coatings such as silver that liberate small amounts of bactericidal ions for extended periods of time; or (iii) the release of embedded bioactive substances from a slowly degrading matrix such as bone cement or hydrolysable organic polymers [16-20]. All strategies have specific advantages depending on the application, and also serious side effects which highlight the requirement for further research [21-23]. After getting into contact with blood or other fluids present in the wound covalently bound molecules may rapidly be buried and rendered inactive by a layer of proteins and other molecules that spontaneously adhere to the implant surface. This appears to be the case with silver-containing coatings that in vivo are less effective than expected; in addition, silver ions exhibit significant tissue toxicity [24, 25]. Bone cement with antibiotic additions may deteriorate and acidic polymer degradation products can cause local inflammation [10]. Madurahydroxylactone (MHL), a polysubstituted benzo[a]naphthacenequinone, is a secondary metabolite that is produced as a mixture of related substances by some strains of Nonomuraearubra. The red pigmented mixture called maduramycin has been reported to be active against gram-positive bacteria, [26, 27]. Even though MHL was discovered earlier, the correct structure was identified by X-ray crystallography only in 1994 [26, 28]. Purified MHL has a marginal antibacterial activity, it is insoluble in physiological salt solutions but readily soluble in DMSO or alcohols, and in alkaline aqueous solutions where the lactone ring opens and maduranic acid salts can form (Fig. 1A upper right) [29]. To investigate the basic feasibility and the release characteristics MHL coatings were generated. After drying of a precursor solution coatings were obtained on various medical implant materials that were firm and biocompatible in cell culture assays. With the addition of a conventional antibiotic effective antibacterial activity was observed that gradually decreased in physiological aqueous solutions within a few days. Strategies are discussed that may further prolong the useful time period of the coating.
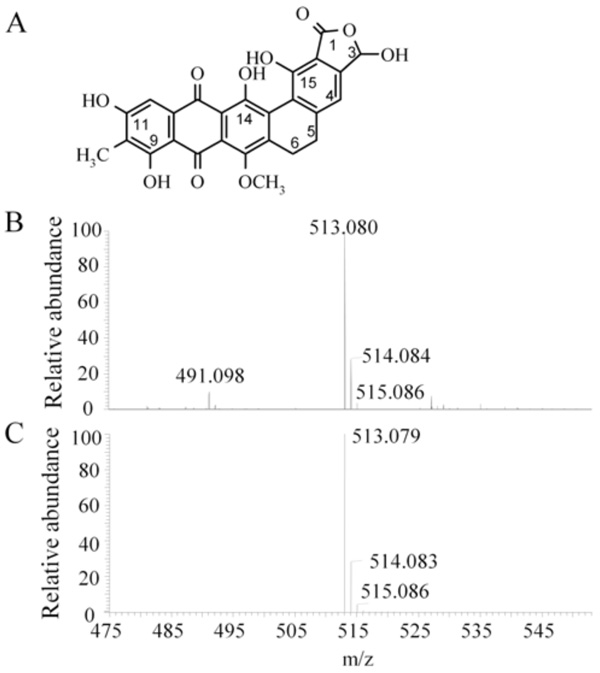
Chemical structure and mass spectrometric analysis of madurahydroxylactone. Chemical structure of MHL (3,9,11,14,15-pentahydroxy-7-methoxy-10-methyl-1,3,5,6,8,13-hexahydronaphthaceno [1,2-f] isobenzofuran-1,8,13-trione) [28]. (A). Electrospray ionization mass spectrometry of the madurahydroxylactone preparation used throughout the described experiments (B) and the simulated mass spectrum for [C26H18O10Na]+ (C) corresponding to the structure depicted in (A).
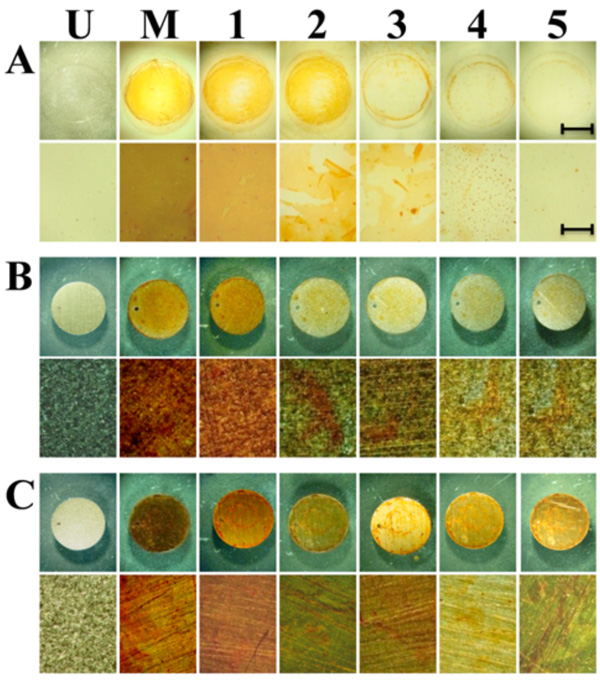
Gradual release of madurahydroxylactone coatings from glass and metallic implant materials. Disks of glass (A), titanium (B) and stainless steel (C) were coated by placing the disks on a hot plate at 90°C. 100 µl of 1 mg/ ml solution of madurahydroxylactone in 70% ethanol was added. After the solvent was evaporated the disks were incubated in PBS solution and pictures of the surface were taken each day until day 5 (1 to 5) at magnifications of 0.6x (top rows) and 6.6x magnification (lower rows). The scale bars correspond to 5 mm (top rows) and 0.5 mm (lower rows) respectively. U, uncoated samples; M, Madurahydroxylactone coated samples after drying.
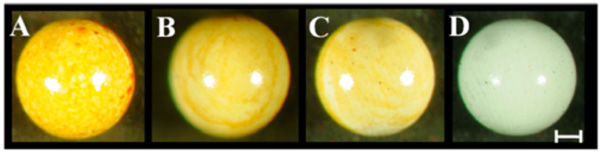
Robust madurahydroxylactone coatings on zirconium beads. Zirconium dioxide beads of 1mm diameter were dip coated using madurahydroxylactone dissolved in DMSO at 100 mg/ml (A), 10mg/ml (B), 1mg/ml (C), DMSO (D) and dried at ambient temperature. The scale bar corresponds to 0.2 mm.
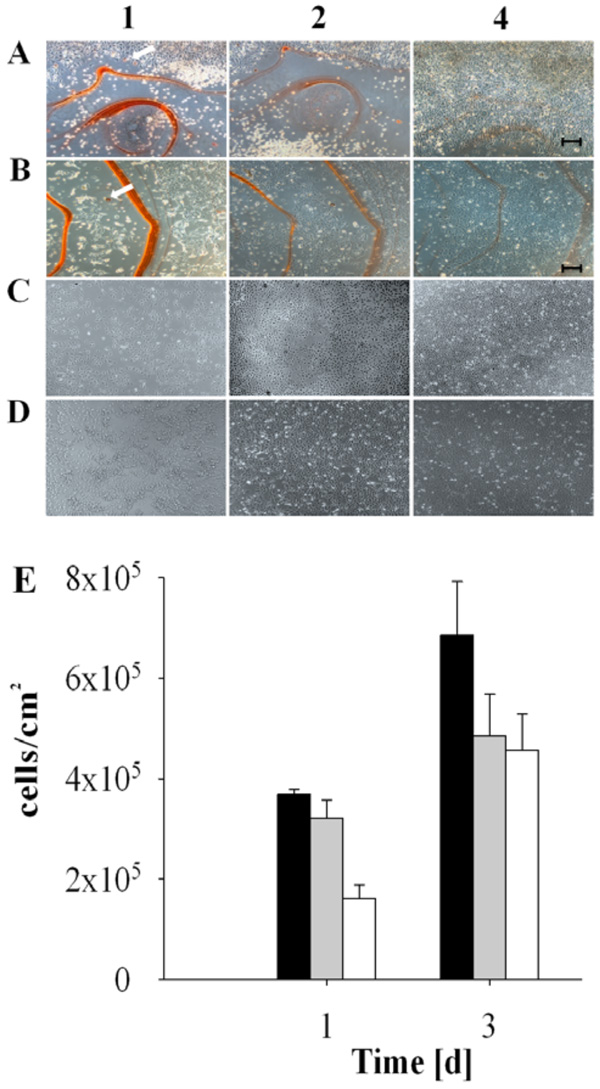
Biocompatibility and delayed adherence of cultured cells on madurahydroxylactone coatings. NIH3T3 cells (A and C) and HEK 293 cells (B and D) were seeded on glass microscope cover slips coated with a 1:1 (w/w) mixture of madurahydroxylactone with ciprofloxacin (A and B) or without coating (C and D). The cells were cultured in DMEM with 10% FCS under standard conditions and microphotography images were taken after 1, 2 or 4 days as indicated. The scale bar corresponds to 100 µm. The white arrows point out red stained cells. HEK 293 cell density after incubation for the time indicated on the following surfaces: CCP (black bars), glass (gray bars), MHL (empty bars) (E).
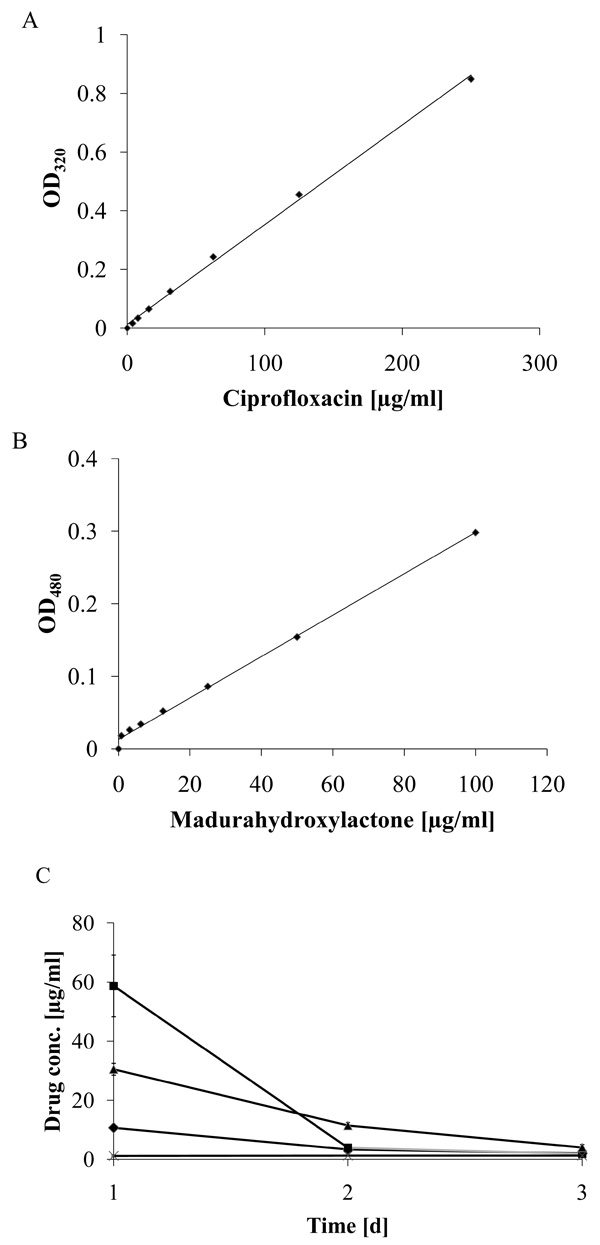
Delayed release of ciprofloxacin from madurahydroxylactone coatings. The specific absorption of ciprofloxacin (A) and madurahydroxylactone (B) in PBS at 320 nm and 480 nm, respectively, was determined by a linear regression analysis. Time dependent ciprofloxacin release from zirconium bead coatings with ciprofloxacin (squares); release from mixed coatings containing ciprofloxacin and madurahydroxylactone (triangles) or from the dried solvent DMSO (crosses); release of madurahydroxylactone from madurahydroxylactone coatings (diamonds). The coated beads were incubated in PBS at 37°C. The buffer was exchanged each day and the OD480 and OD320 determined to calculate the release of ciprofloxacin and madurahydroxylactone, respectively (C).
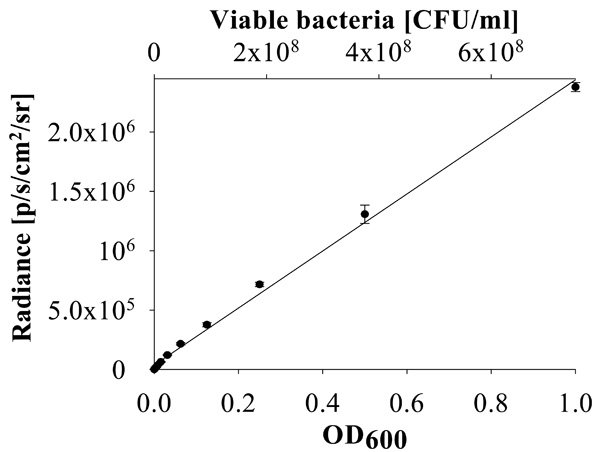
Linear relationship between bacterial concentration and luminescence. Correlation of P. aeruginosa PA01 CTX::lux strain luminescence (radiance) and OD600 of the culture with the titer of bacterial colony forming units (CFU/ml).
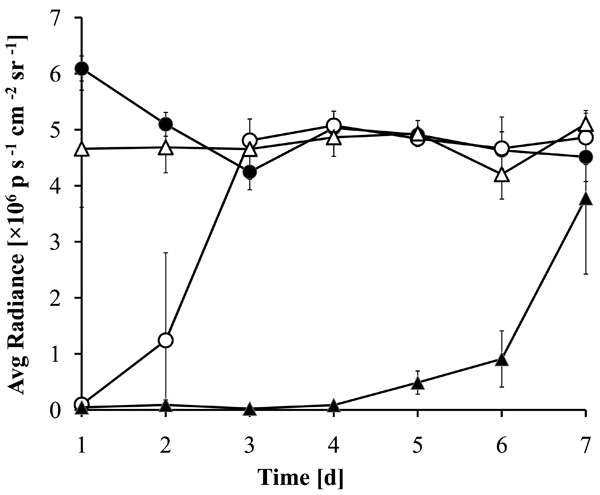
Prolonged antibacterial activity of MHL coatings with ciprofloxacin. Zirconium dioxide beads coated with a 1:1 (w/w) mixture of MHL with ciprofloxacin were incubated for 7 days in LB medium with daily exchange of the medium. The medium was inoculated with luminescent P. aeruginosa and the luminescence of the cultures after 5 hrs incubation at 37°C is shown. The supernatants were incubated with beads coated with MHL (filled circles), ciprofloxacin (open circles), DMSO (open triangles) or a combination of ciprofloxacin with MHL (filled triangles).
MATERIALS AND METHODS
Madurahydroxylactone Preparation
The secondary metabolite MHL was produced biotechnologically by cultures of the Gram-positive soil bacterium Nonomuraearubra (ATCC 27031, DSM 43768T, IMET13001, 16S rDNA: AF277200; basonyms Nonomuria rubra, Actinomadura rubra, Microtetraspora rubra, Micromonospora rubra [30-33]. MHL was extracted and purified using high-performance liquid chromatography (HPLC) as described [26-28]. The preparation was stored at -20 ºC as a dry powder or dissolved in DMSO until it was used.
ESI-MS Analysis of MHL
The sample was dried and dissolved in methanol (ca. 3 pmol/µl). An Orbitrap/Velos mass spectrometer (Thermo Scientific, Bremen, Germany) equipped with a nanospray ion source was used. The spectrum was recorded in the positive mode at a resolution of 100 000 and compared with the simulated spectrum generated by the Xcalibur (Thermo Scientific, USA) software (Fig. 1B and 1C).
Coating Procedures
For biocompatibility testing coatings were prepared as follows. A 10 mg/ml MHL solution in dimethyl sulphoxide (DMSO) was prepared. Ciprofloxacin (Fluka Chemie GmbH, Deisenhofen, Germany) was not soluble in DMSO; therefore a fine suspension was prepared at a concentration of 10 mg/ml ciprofloxacin in DMSO. The wells of a 12-well tissue culture polystyrene (TCPS) plate (BD Falcon, BD Biosciences, Germany) were coated with 1µl each of the MHL solution or with a 1:1 (w/w) mixture of MHL and ciprofloxacin. The coatings were dried overnight at room temperature. Cells were seeded directly on these coatings described below. Zirconium dioxide (zirconia) beads of 1 mm in diameter (BioSpec Products, Inc., Bartlesville, OK, USA) were dip-coated, followed by drying overnight at room temperature. Beads dipped in the solvent DMSO were used as a control. The release of ciprofloxacin from the coatings was determined by submerging the beads in 10µl of 0.1 M phosphate buffered saline (PBS, pH 7.4) in a microcentrifuge tube (Eppendorf, Germany) and kept at 37°C. The liquid was removed at 24h intervals and fresh PBS was added to the beads. The resulting ciprofloxacin concentration in the supernatants was determined by measuring the absorbance at 320nm using a NanoDrop ND-1000 UV-Vis Spectrophotometer (NanoDrop Technologies, Wilmington, USA). MHL released from the coatings was calculated similarly by determination of the OD480 of the sample supernatant. Titanium, stainless steel (brushed) and glass discs of 9mm diameter were coated with a single layer of pure MHL dissolved in 70% ethanol at a concentration of 1 mg/ ml. The discs were put on a hot plate that was set to 90ºC and the plate surface was protected by a household aluminum foil. 100 µl MHL solution was added with a pipette and spread out quickly with the pipette tip until the surface was completely covered. After drying the coatings were incubated in phosphate buffered saline (PBS) solution as described in the text and photographed using a STEMI SV11 stereo zoom-microscope (Carl Zeiss, Oberkochen, Germany) equipped with a digital camera DSC-S75 (Sony, Germany).
Bacteria
The Escherichia coli laboratory strains Top 10 (F– mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara leu) 7697 galU galK rpsL (StrR) endA1 nupG) and DH10B (F– mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara leu) 7697 galU galK rpsL nupG λ–), were purchased from Invitrogen (Germany). Pseudomonas aeruginosa SCV 20265 is a small colony variant with increased antibiotic resistance that was originally isolated from the lung of a cystic fibrosis patient [34, 35]. Pseudomonas aeruginosa strain PA01 CTX::lux is a laboratory strain that has been made luminescent by the insertion of the entire lux operon encoding sequence into the genomic DNA [36, 37]. The strains S. aureus COL and MRSA are both methicillin resistant and their genomes have been completely sequenced [38-44]. Bacteria were grown on LB agar dishes or as suspension cultures in LB broth according to standard procedures [45]. Bacterial growth in suspension cultures was monitored by measuring the optical density at 600 nm (OD600). The relative bioluminescence of P. aeruginosa PAO1 CTX::lux in suspension versus the optical density (OD600) was determined by serial dilutions of a fresh overnight culture in 100µl LB in a 96-well plate at 37º C using an optical in vivo imaging system (IVIS-200, Xenogen, Alameda, CA).
The antibacterial effects of MHL on solid media were scored by a radial streak assay. 50µg MHL dissolved in DMSO (10mg/ml) were placed in the center of a Petri dish with LB agar. For comparison, ciprofloxacin (50µg) and a combination of 25µg MHL and 25 µg ciprofloxacin and 5µl of the pure solvent DMSO were put on separate Petri dishes. After drying freshly diluted overnight cultures of bacteria were radially streaked from the outer rim towards the center of the dish and the dishes incubated overnight at 37ºC. The antibacterial efficacy was scored by measuring the distance of the growth inhibitory zone from the center (Table 1). The antibacterial activity released from MHL and ciprofloxacin coated zirconia beads was assayed by submerging the beads in 50µl of LB in a microcentrifuge tube at 37°C. The supernatants were collected at 24h intervals and replaced with 50µl of fresh medium. 45µl of each supernatant was inoculated in a 96 well plate with 5µl of a PAO1 CTX::lux culture at an OD600 of 0.2. At the times indicated the bacterial luminescence from each sample was determined using an IVIS-200 system. To determine the titer of viable bacteria aliquots of serial dilutions from each of the supernatants were spread on LB agar plates and after overnight incubation at 37°C the colonies were counted. All experiments were performed in triplicates and the data is presented as average value with the standard deviation.
Bacterial Growth Inhibition by Madurahydroxylactone
| Bacterial strains | Madurahydroxylactone inhibition zone [mm] | Ciprofloxacin Inhibition zone [mm] | Madurahydroxylactone + ciprofloxacin inhibition zone [mm] | DMSO inhibition zone [mm] |
|---|---|---|---|---|
| Staphylococcus aureus (Col) | 1 | 4.5 | 6 | 0 |
| Staphylococcus aureus (MRSA) | 1 | 4.5 | 4.5 | 0 |
| Escherichia coli (Top 10) | 3.5 | 9 | 8 | 0 |
| Escherichia coli (DH10B) | 3 | 5 | 4.5 | 0 |
| Pseudomonas aeruginosa (SCV20265) | 0 | 6 | 4.5 | 0 |
| Pseudomonas aeruginosa (PAO1 CTX::lux) | 0 | 9 | 6 | 0 |
Mammalian Cells
The murine fibroblast cell line NIH 3T3 (ATCC CRL-1658) and human embryonic kidney cell line HEK 293 (ATCC CRL-1573) were stored in liquid nitrogen and cultured according to standard procedures as previously described [46]. NIH 3T3 cells were cultured in Dulbecco’s modified Eagle’s Medium (DMEM; Gibco/BRL), supplemented with 10% (v/v) fetal calf serum, 1% (v/v) glutamine, and 1% (v/v) penicillin and streptomycin (100 U/ml penicillin G and 100 µg/ml streptomycin) at 37°C in a humidified atmosphere with 5% CO2. A near confluent culture of NIH 3T3 cells was detached using a trypsin solution according to standard procedures and diluted 1:5 in fresh cell culture medium. 2ml of the cell suspension was added to each of the coated wells. After incubating the cells in a humidified atmosphere with 5% CO2 at 37°C for 24, 48, or 96 h, respectively, the cells were examined using a Zeiss Axio Observer A-1 inverted visible light microscope and images were recorded using the Axiovision Rel. 4.5 software (Carl Zeiss MicroImaging Inc., Germany). The same procedure was used for evaluating the biocompatibility of MHL coatings with HEK 293 cells, except that the culture medium used was minimum essential medium (MEM; Gibco/BRL) supplemented with 10% fetal calf serum, 1% glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin. All experiments were conducted in triplicates and the results are given as mean values with the standard deviation.
RESULTS
Confirmation of the Purity and Molecular Mass of the MHL Preparation
To confirm the purity and the molecular weight of MHL as calculated by its chemical structure (Fig. 1A), the dark red colored preparation was characterized by electrospray ionization mass spectrometry (ESI-MS). An excess of sodium ions were added to a methanolic solution to obtain a homogeneous sodium salt preparation and to suppress mass peaks of other salts. The most abundant molecular weight species with a mass of 513.080 agrees well with the calculated molecular weight of 513,079 of the sodium adduct of MHL (Fig. 1B and C). Minor peaks at 514.084 and 515,086 correspond to the natural relative abundance and to the calculated molecular weight of the compound with one or two C12 carbon atoms replaced by the C13 isotope, respectively. The peak at m/z 491.098 corresponds to the protonated form of the molecule. The nature of the remaining mass peaks of substances of minor abundance in the preparation was not further investigated. The analysis showed that the preparation used was pure and homogeneous and in agreement with the corrected MHL structure [28].
Preparation of MHL Coatings on Implant Materials
For coating microscope cover slips MHL was dissolved in DMSO and the solution was dispersed on the glass surface. Upon drying a readily visible orange-red colored MHL film formed. During drying, more material was deposited on the outer rims of the liquid covered area. There were no cracks or holes in the resulting coating. Whereas spray coating turned out to be comparatively wasteful, a more equal distribution was achieved when a MHL solution in ethanol was applied with a pipette onto preheated material samples. With this technique the most widely used metallic implant materials in the clinic, surgical steel and titanium, could be coated with a MHL layer with a more homogeneous appearance (Fig. 2; M). As an example for a ceramic implant material, zirconium dioxide is widely used for tooth repair or as replacement prostheses. ZrO2 beads were coated by dip coating in a MHL solution in DMSO followed by drying at ambient temperatures. The thickness of the resulting layer was dependent on the MHL concentration in the precursor solution; however, all coatings were firmly attached and had a homogeneous, shiny and smooth appearance (Fig. 3). This indicates that curved ceramic implant surfaces can be coated with a firm layer of MHL.
Biocompatibility of MHL Coatings
The biocompatibility of MHL coatings was investigated by monitoring cultures of human and murine cells seeded directly on MHL coated glass slides. Cell attachment, cell spreading and cell proliferation were evaluated microscopically at various time points. Initially cells did not attach to MHL coatings, the cells remained rounded and in suspension. A few individual rounded cells showed an intense red staining (Fig. 4, arrows). 4 days after seeding both cell types investigated eventually adhered to the coating and showed a spread out physiology (Fig. 4A and 4B). Similar to the control, the cells then proliferated on the MHL layer until confluence was reached (Fig. 4E). This shows that MHL coatings are biocompatible in cell culture assays and after some delay they can even serve as a substrate for mammalian cell adhesion.
Antibacterial Efficacy of MHL
MHL has been reported to have antimicrobial effects against Gram-positive staining bacteria [29]. The antibacterial efficacy of MHL was examined with a plate assay using Gram-positive S. aureus as well as Gram- negative E. coli laboratory strains and P. aeruginosa, a drug-resistant biofilm forming pathogen. All bacterial strains tested were highly resistant to MHL in plate assays (Table 1). Since MHL showed promising properties as a slow release coating but only limited effects against bacteria, combinations with a well established antibiotic with proven activity were used to increase the antibacterial efficacy of the coatings. Among various MHL to ciprofloxacin ratios tested, the 1:1 (w/w) mixture formed a well adhering coating and was highly effective against all bacterial strains tested (Table 1). Therefore, it was selected for further investigations.
Release Characteristics of MHL Coatings
The coating remained attached to these materials even after immersion in a phosphate buffered solution (Fig. 2). To determine the stability of the coatings under physiological conditions the MHL coated implant material discs were incubated in PBS at 37°C and 5% CO2 atmosphere in a cell culture incubator with a daily exchange of the supernatant. The single layer coatings gradually dissolved in the course of one to two weeks while remaining attached to the metal surface (Fig. 2). In contrast, under these conditions flakes of the coating peeled off from the glass surfaces that were used here as controls, indicating a less firm attachment to this material. These results show that MHL fulfills some basic criteria for applications as a slow release coating for metallic implants with a time course that limits its suitability to some specific short-term applications.
To determine the characteristics of the coating on zirconium dioxide beads these were incubated in a physiological phosphate buffer, whereby the buffer was exchanged at daily intervals. The ciprofloxacin and MHL concentrations in the supernatant could be determined by measuring the absorption at 320 nm and 480 nm, respectively, with a nearly linear correlation of the optical density and the concentration within the range investigated (Fig. 5A and 5B). The release of pure MHL from coatings was even slower whereas ciprofloxacin from coatings without MHL was largely dissolved in one initial burst on the first day (Fig. 5C). The concentration of ciprofloxacin released from the mixed coating with MHL was highest in the first day of the experiment and decreased gradually until day 3 when the absorption reached background levels and was no longer detectable. Therefore, for measuring the small amounts released after longer incubation periods the absorption measurements were not sensitive enough.
To establish a more sensitive antibiotic release scoring method genetically engineered luminescent Pseudomonas aeruginosa bacteria were used. This allowed the contact-free quantification of the antibacterial effects in small assay volumes. The luminescence was linear proportional to the viable bacterial cell density over a wide range of several orders of magnitude (Fig. 6). Therefore the luminescence could be directly used as a measure of cell growth and viability. Using this assay the supernatant from the beads coated with a 1:1 (w/w) mixture of MHL and ciprofloxacin significantly inhibited bacterial proliferation until day 7 (Fig. 7). The highest efficacy was observed during the first day, with a gradual decrease over the time of the experiment and resulting in a marginal antibacterial effect at day 7. In contrast, the bactericidal effect of a pure ciprofloxacin coating was reduced over 90% after one day incubation. This is in good agreement with the absorption measurements of the supernatant of the incubation buffer, showing that this test was suitable and sufficiently sensitive. The results indicated that the combined coating of ciprofloxacin with MHL was gradually released when exposed to an aqueous environment.
DISCUSSION
Structurally related derivatives of MHL have been synthesized with an enhanced antimicrobial activity and were proposed as slow release antibiotic coatings for medical implants [29, 47]. However, the efficacy of these compounds was limited. A test using luminescently labeled bacteria was established for real-time monitoring of the antimicrobial activity of novel implant coatings. The advantages of this assay are that unlike microscopic techniques, it requires no staining procedures or other sample preparation, it works with opaque or fluorescent implant materials, it yields quantitative numerical data of the same samples over time and when using microtiter plates several hundred samples can be evaluated contactless, in parallel in a single measurement. Our data shows that MHL is a chemical compound with properties that appear suitable as a basis for implant coatings. MHL coatings did not peel off; they remained bound to the implant surface and were gradually dissolved in physiologically buffered aqueous solutions. MHL was highly biocompatible in cell culture assays, which is consistent with the low toxicity observed in mice with a reported LC50 greater than 250 mg/kg intraperitoneally [26]. The fact that cultured cells adhere to MHL coatings with a delay is not necessarily related to biocompatibility. A possible explanation would be that the cell culture medium exposed MHL surface layer may change its characteristics by absorbing lipophilic molecules such as serum albumin [26]. The finding that initially individual floating cells at the border of the coating appeared intensively red stained suggests that these cells were in close contact with the MHL layer and visible amounts of MHL were bound to the cell membrane or were taken up by these cells. A highly biocompatible behavior may be of advantage compared to some of the presently used slow release implant materials. In particular, various degradable organic polymers have been reported to cause inflammation and even bone cement degradation may result in the accumulation of inflammatory particulate debris [48-52]. It has been demonstrated that drug combinations with ciprofloxacin can have synergistic effects on implant associated biofilms of the major clinically relevant bacterial species [53]. Therefore, we prepared MHL coatings with additions of ciprofloxacin which remained efficacious for a few days in aqueous solutions. This time period is within the range that has been observed with related coating techniques and appears promising to prevent nosocomial infections [54-56]. However, an extension of the effective time period could be of advantage, since implant infections have been reported to occasionally occur not only during surgery but even well after the wound has healed, possibly by pathogens that are disseminated in the body via the blood circulation [57]. There are various strategies by which the effective time span of the coating could be further extended, for example by applying thicker layers, by multilayered coatings or by loading the antibiotic into the cavities of implants with porous surface structures [47, 58, 59].
CONCLUSION
MHL was used as a basis to generate antimicrobial delayed drug release coatings on various medical implant materials. A luminescent bacteria assay has been established that facilitated the evaluation of the antimicrobial efficacy. The results show that MHL-based coatings have promising physical and biological properties to prevent nosocomial implant infections at least during the most risky period up to one week after surgery.
ACKNOWLEDGEMENTS
We thank Susanne Häussler and Eva Medina for the gift of bacterial strains and Herbert Weich for providing laboratory space. This research was funded by the German Research Foundation - Collaborative Research Centre 599: “Biomedical Technology”. M.B. was supported by a DAAD Scholarship For Doctorate Studies, by the HEC-DAAD Fellowship Programme.

