All published articles of this journal are available on ScienceDirect.

A Review of the Design Process for Implantable Orthopedic Medical Devices
Authors Info & Affiliations
Abstract
The design process for medical devices is highly regulated to ensure the safety of patients. This paper will present a review of the design process for implantable orthopedic medical devices. It will cover the main stages of feasibility, design reviews, design, design verification, manufacture, design validation, design transfer and design changes.
1. INTRODUCTION
The design process for medical devices is highly regulated to ensure the safety of patients and healthcare workers. The Medical Device Directive [1] was developed to regulate medical devices in Europe. It is a document that is legally binding, enforceable in law and with penalties for non-compliance. Regulations outside Europe vary. For example, in the United States of America, the Food and Drug Administration (FDA) is responsible for the safety of medical devices [2]. In order to comply with the regulations, companies are required to have a quality management system in place [3-5] to ensure that the whole design process is managed and planned in a systematic and repeatable manner. To show compliance with the regulatory aspects it is necessary to maintain a Design History File (which can also be know as a Technical File or Design Dossier) which describes the design history of a product and is maintained post-product release to include subsequent changes to the product and relevant post-market surveillance data. The aim of this paper is to give an overview of the design process for implantable orthopedic medical devices.
2. DESIGN PROCESS
2.1. Overview
The medical device design process, as with other design processes, can be broadly divided into six areas [6-9]:
- Market
- Design specification
- Concept design
- Detail design
- Manufacture
- Sell
However, in this paper we propose a more detailed structure to the design process, shown in Fig. (1). Each of these aspects of the design process will be discussed in the following sections.
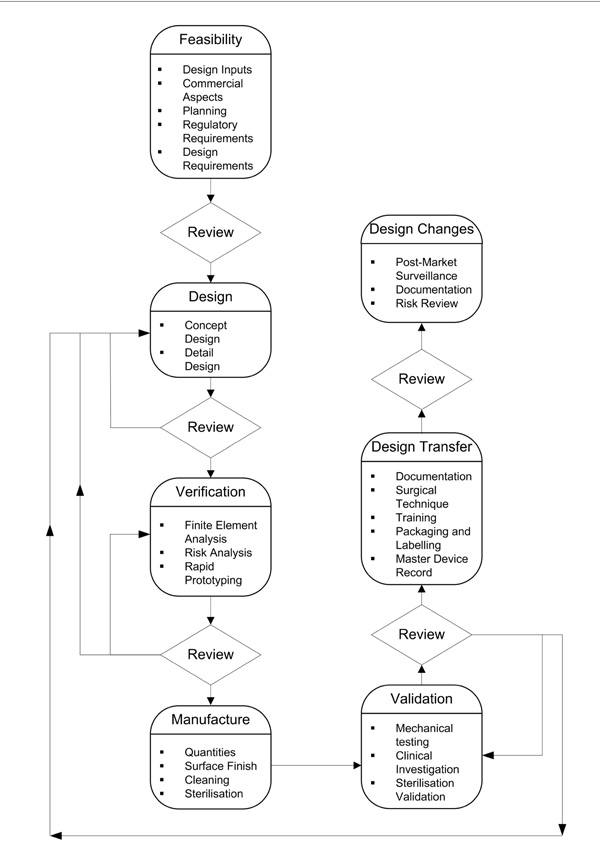
The medical device design process.
2.2. Feasibility
2.2.1. Design Inputs
Ideas for new or improved medical devices will come from surgeons, other clinicians, sales teams or medical engineers, perhaps working together as an interdisciplinary team. At the initiation of the project the basic objectives and clinical indications for the device will be defined. A surgical review panel may be formed to help guide the design and ensure that the device functions correctly surgically and that any instruments are suitable for use by the surgeon.
2.2.2. Commercial Aspects
There has to be a market or customer need for a medical device to be designed and produced. It is not commercially viable to design a device that has a limited market, whether this is due to competition from well established products or from a limited numbers of patients. A feasibility study for a new idea for a device needs to be undertaken to identify the potential market share, similar devices produced by competitor companies and the potential market value of devices. A review of the intellectual property is also required to determine whether the design can be protected and does not infringe other patents.
2.2.3. Planning
It is important that each aspect of the design process is project managed with achievable milestones set and defined throughout the project life cycle. The setting of milestones should enable the project manager to assess the progress of the project to ensure that it is completed on time and within budget. The definition of the milestones should detail the requirements for the project to pass the milestone, for example completing the market feasibility report and the project objectives. The project plan should include regular milestones and project meetings to assist with the identification of problems and allow early action to be taken to counteract delays to a project. The planning process should also include a risk management plan, which should highlight the relevant strategies that will be employed to reduce and manage the risks that are associated with a project. Finally the planning process should identify the human and financial resources required to be in place to successfully realise a design.
2.2.4. Regulatory Requirements
All medical devices must have regulatory approval before they can be released to market. There are a number of internationally and nationally agreed requirements to which medical devices must conform. Currently these standards are not harmonized, and there are differences between those that regulate Europe and the United States. Medical devices must also be classified according to the level of risk associated with their use upon a patient and to the user. The classification process of a medical device will determine the relevant approval route to market. It is therefore essential that the regulations that will influence the design of a product are identified early in the design process. Independent advice on this area must be sought from a Regulatory Authority, Conformity Assessment Body or other authorised third party.
In the design of the product it is necessary to identify global standards, which apply to all medical devices, semi-global standards which apply to a particular family of devices and specific standards that apply to particular devices or pieces of equipment. By identifying these standards complications in the latter stages of the design process can be avoided. Whilst not all of the standards are mandatory, compliance will often help to accelerate the approval process.
2.2.5. Design Requirements
The design requirements (or product design specification) are essential before a medical device can be designed. It sets out exactly what is required of the design against which each stage of the design can be verified. Whilst the initiation of the project will highlight some of the technical requirements for a project, it is important to perform a thorough requirement capture process to ensure that all of the functional performance needs are identified early in the design process. It is important to remember that the requirements are solution independent to prevent narrowing the design options available. A general standard exists to help determine the design requirements [10]. Many types of medical device also have particular requirements, such as joint replacement implants [11] and implants for osteosynthesis [12]. In addition, specific requirements can be specified for certain devices, such as joint replacement implants for the hip and knee [13,14].
Most implants require custom designed surgical instruments and it is vital to consider the product design specification for the instruments at an early stage as they interact with the implant [15]. Any design changes to the implant will have a knock-on effect for the design of the instrument. In many cases the complexity and design time of the surgical instruments are greater than the actual implant itself. The standard BS EN 12011 [16] has been produced to help identify the requirements for surgical instruments. It is also important to consider the packaging, sterilization [17] and labeling [18] at an early stage, as these are an integral part of the whole device. The design requirements for the device will include:
- intended performance
- design attributes
- materials
- design evaluation
- manufacture
- testing
- instruments required
- sterilization
- packaging
- information to be supplied by the manufacturer
The design requirements vary between orthopedic devices. For a total knee arthroplasty the important requirements would be for the implant to last for 25 years, have the required range of motion, prevent loosening and minimise wear debris [19]. In the design of a new wrist arthroplasty it would be important to consider the range of motion (flexion/extension, radial/ulnar deviation or radiocarpal rotation), fixation and materials [20]. For the design of an intramedullary nail stabilising the bone while the fracture heals would be an important requirement [21].
2.3. Design Reviews
A design review is required, at each stage of the design process, to formally document comprehensive, systematic examination of a design to:
- evaluate design requirements
- assess capability of the design
- identify problems
2.4. Design
2.4.1. Concept Design
The concept, or conceptual, design stage is where solutions are generated to meet the design requirements. The aim is to generate as many ideas as possible. At this stage ideas should not be judged. Various methods exist to help with creativity in developing concept designs such as Brain storming [22-24] and TRIZ (Theory of Inventive Problem Solving) [22]. Concept design may involve:
- simple sketches of ideas;
- computer aided design models;
- analytical calculations;
- initial manufacturer consultation.
Once a range of concept designs have been developed it is worth assessing each of the concept designs for patentable technology to ensure that the final design has been fully protected. The protection of ideas and concepts can play an important role in the success of a product once launched to the market. It is therefore essential to consider this as early as possible in the design phase.
The range of concept designs can then be systematically rated by the design team to determine the most suitable concept to develop, in the detail design stage. A variety of methods for concept selection exist such as the Six Thinking Hats or the use of matrices where concepts are scored against a set of criteria that is required of the design [6,7,23,25].
2.4.2. Detail Design
At the detail design stage the flesh is put onto the bones of the chosen conceptual idea [7]. A concept design is worked through until a detail design has been produced. This will include:
- generation of solid computer aided design models
- specification of materials
- drafting of engineering drawings
- analysis of tolerance stacks within associated assemblies to ensure correct operation
- detailing with the inspection requirements to ensure that the part/assembly operates correctly
- liaison with manufacturers to ensure that the device is designed for manufacture (DFM), designed for assembly (DFA) or designed for manufacture and assembly (DFMA)
2.5. Design Verification
2.5.1. Introduction
Design verification involves confirmation by examination that a medical device meets the design requirements and is essentially asking the question “are we building the thing right?” [26,27]. It is essential that verification is considered early in the design process. Ideally, when a design requirement is decided, a method for verifying that requirement should also be developed [27]. Design verification methods can include:
- finite element analysis
- risk analysis
- rapid prototyping
2.5.2. Finite Element Analysis
Finite element analysis is a widely used technique in medical device design and can be used to verify if a design will have sufficient strength to withstand the loading conditions in the human body [28]. Finite element analysis is a proven cost saving tool and can reduce design cycle time. In the development of a new method for closing a median sternotomy using cannulated screws and wire, finite element analysis was used to investigate the stresses acting between the screw and sternum [29]. In knee replacement implants, finite element analysis can be used to optimise the bearing surfaces [30]. Finite element analysis can also be used to investigate design changes to existing devices. For example, Mathias et al. [31] used finite element analysis to investigate the effect of introducing holes into the femoral component of a total hip replacement implant to engage with a stem introducer instrument and Leahy et al. [15] undertook a redesign of a flexible fixation system for the lumbar spine based on an existing design.
2.5.3. Risk Analysis
A key part of the medical device design process is to undertake a risk analysis [32]. Any medical device should be designed and manufactured so that it does not compromise the safety of patients or healthcare workers. Manufacturers must eliminate or reduce risks as far as possible; any risks that exist must be weighed against the benefits to the patient. The way to show risks have been eliminated or reduced is to undertake a risk analysis. Many techniques, such as Failure Mode Effect Analysis and Fault Tree Analysis can be used in a risk analysis; guidance for these methods can be found in standards [33] and [34], respectively. Failure Mode Effects Analysis (FMEA) can have a number of variants which address different aspects of the product development cycle, all of which use a bottom-up approach to evaluate risks associated with aspects of the design. Examples of the variants are Design Failure Mode Effects Analysis (DFMEA), Process Failure Mode Effects Analysis (PFMEA), Failure Mode Criticality Analysis (FMECA) and User Failure Mode Effects Analysis (UFMEA). The aim of these methods is to consider all the possible risks associated with a medical device and identify ways that these risks can be eliminated or reduced. In FMEA the occurrence, severity, and detection of each failure are rated on a scale from 1 to 10. A risk priority number is then calculated by multiplying the three ratings together. The design team must then decide if a possible failure mode is acceptable or if ways need to be found to reduce it. A partial risk analysis for a Swanson wrist implant is shown in Table 1 [32].
Partial Results of a Risk Analysis for the Swanson Wrist Implant. The Occurrence (O), Severity (S) and Detection (D) are Rated on a Scale of 1 to 10 and Multiplied Together to Give a Risk Priority Number (RPN)
| Possible Failure Mode | Effect of Failure | Cause of Failure | O | S | D | RPN | Action to Reduce or Eliminate Risk |
|---|---|---|---|---|---|---|---|
| Breaks | Device does not function as intended | Fatigue failure | 10 | 6 | 5 | 300 | |
| Tears from sharp edges of bone | 10 | 6 | 5 | 300 | Grommets have been designed for use with the Swanson wrist implant | ||
| Damaged during implantation using sharp instruments | 2 | 6 | 5 | 60 | Use of correct instrumentation; trained hand surgeon; adequate and clear instructions for use | ||
| Incorrect implantation technique | 2 | 6 | 5 | 60 | Trained hand surgeon; adequate instructions for use | ||
| Wear particles of silicone | Silicone synovitis | Implant rubbing against bone | 8 | 6 | 7 | 336 |
It is also important not to isolate the medical device from the surgical instruments, packaging, sterilization and labeling; these have many risks associated with them that could adversely affect the performance of a medical device. An international standard for medical device risk analysis has been published [35]. Risk analysis helps to realise a design if it is undertaken at an early stage and should be undertaken at stages during the design process, including a final risk analysis.
2.5.4. Rapid Prototyping
Rapid prototyping is a very effect technique for verifying the design of medical devices as it aids communication between engineers and surgeons [36]. Models of implants can be produced within hours by a variety of methods such as Selective Laser Sintering (SLS), Fused Deposition Modeling (FDM) or three-dimensional printing [37]. The surgeon can then inspect the models and size them against a skeleton. Rapid prototyping can also be used to produce models of human bones, which is particular useful if there is abnormal anatomy into which an implant will be used [38]. Multi-component systems can also benefit from the use of rapid prototyping as they allow designers to evaluate the interaction between components, such as a screw and a screw driver, thus minimising the opportunity for component incompatibility.
2.6. Manufacture
Before the design is transferred to production it is essential to ensure that the chosen manufacturing processes are repeatable and reliable. The choice of manufacturing technique depends on many factors:
- number to be produced
- surface finish required
- post machining cleaning processes
- sterilization process (if necessary)
As well as the manufacture of the devices and the surgical instruments required to implant the device, packaging for the device and instruments, sterilization techniques, operation instructions and labeling printing requirements also need to be finalised.
2.7. Design Validation
Validation of the device is performed under actual or simulated conditions for use. While verification is answering the question “are we building the thing right”, validation is asking “have we built the right thing” [26,27]. Validation is to ensure that the medical device meets the user requirements and the intended use. Validation can include:
- mechanical testing of prototypes
- evidence that similar medical devices are clinically safe
- a clinical investigation
- sterilization validation
Mechanical testing allows the mechanical conditions in the human body to be simulated in the laboratory. Mechanical testing will give no indication of how the device will behave in the body from a biological point of view, but it will ensure that devices have sufficient strength and stiffness to perform as required. There are a large number of standards available to guide the pre-clinical mechanical testing of joint replacement implants. For a total hip joint replacement it would be necessary to:
- determine the endurance properties of the stem to BS 7251-12 [39] by using a materials testing machine to apply a sinusoidally varying force (between 300 N and 2300 N) for up to 5 million cycles;
- to determine the wear of the bearing surfaces to BS ISO 14242-1 [40] by using a hip simulator to subject a hip replacement implant to loads of up to 3000 N and motions similar to those encountered in the body. The amount of wear is determined at 0.5 million and 1 million cycles and then at least every 1 million cycles up to 5 million cycles using either the gravimetric or dimensional methods to determine the wear rate.
In some cases of mechanical testing it is beneficial to use human cadaveric material [41]. For example, cadavers have been used to investigate the attachment of a hook device to the spinous process of the lumbar spine in terms of strength and slippage [42,43]. In addition, cadavers can be used to trial surgical instruments [44].
Once pre-clinical testing has been completed manufacturers of joint replacement implants are required to make a decision as to whether a clinical investigation is required. This is guided by the standard BS EN ISO 14155-1 [45]. A critical review of the literature is required to ascertain similar implants and how they have performed in patients. Analogy may be used to justify not undertaking a clinical investigation. If the manufacturer does decide that a clinical investigation is required a clinical investigation plan must be produced in accordance with BS EN ISO 14155-2 [46]. The investigation will need Ethics Committee approval and the manufacturer will be required to decide on the length of the investigation, the number of patients to be involved and the type of data to be collected.
2.8. Design Transfer
Before a design is transferred to production it is necessary to ensure that all documents and training associated with the device are in place. Design transfer can include:
- generation of instructions for use
- finalization of the surgical technique
- plan the training of surgeons
- finalization of the labeling and packaging
- completed vendor requirements such as audits, first article inspection or surveys
- Total cost bill of materials
- completed inspection plans and process worksheets
- creation of the master device record
2.9. Design Changes
After a medical device is on the market it is necessary for the manufacturer to have a post-market surveillance process in place to ensure the safety of patients and healthcare workers after the device is on the market. Feedback from surgeons may lead to design changes being made to the device or the surgical instruments. The changes that arise from this feedback must be fully documented and the effects upon the device fully investigated. This latter process may involve repeating much of the verification and validation processes, depending upon the magnitude of the change. All design changes must be accompanied by an updated risk assessment to ensure that the full impact of the change has been understood.
ACKNOWLEDGEMENTS
This paper is based on work undertaken during a Knowledge Transfer Partnership (KTP) between the University of Birmingham and Surgicraft Ltd. We would like to thank Dr. David Britton and Ms. Jose Freedman, who acted as KTP Advisors. Funding by Technology Transfer & Innovation Ltd. and Surgicraft Ltd. is gratefully acknowledged.

