All published articles of this journal are available on ScienceDirect.

Amyloid-Negative Dementia in the Elderly is Associated with High Accumulation of Tau in the Temporal Lobes
Authors Info & Affiliations
Abstract
Background:
We previously reported that among cases clinically diagnosed with Alzheimer’s disease, the proportion of amyloid beta (Aβ) -negative case increases in the elderly population. Tauopathy including Argyrophilic Grain Disease (AGD) and Neurofibrillary Tangle-Predominant Dementia (NFTPD), may be the leading causes of such dementia.
Objective:
To evaluate the involvement of tau, we studied tau accumulation in Amyloid-Negative Dementia Cases in the Elderly (ANDE) with Positron Emission Tomography (PET).
Methods:
Seven cases with slowly progressive dementia who were older than 80 years and were negative for Aβ were studied. In one case, autopsy obtained 2 years after the PET examination revealed neurofibrillary tangles limited around the parahippocampal gyrus. Four cases showed strong laterality in magnetic resonance imaging atrophy (clinical AGD), while the other three cases had no significant laterality in atrophy (clinical NFTPD). Age-corrected PET data of healthy controls (HC; n = 12) were used as control. Tau accumulation was evaluated with [11C]PBB3-PET.
Results:
High accumulation was found in the lateral temporal cortex in ANDE. In autopsy case, scattered neurofibrillary tangles were found in the parahippocampal gyrus. In addition, there was a very high accumulation of PBB3 in the large area of bilateral parietal lobes, although no corresponding tau component was found in the autopsied case.
Conclusion:
Relatively high burden of tau deposition was commonly observed in the lateral temporal cortex and parietal cortex of ANDE, part of which may explain dementia in these subjects. [11C]PBB3 may be useful in detecting tauopathy in ANDE.
1. INTRODUCTION
Alzheimer’s Disease (AD) is the leading cause of dementia, and the prevalence of clinically diagnosed AD nearly doubles for every 5 years for those older than 65 years [1-4]. Most of the epidemiological reports are based on the clinical diagnosis of dementia, such as using the criteria from the Diagnostic and Statistical Manual of Mental Disorders (third and fourth editions) and the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s disease and Related Disorders Association [1, 5-8].
| Case | Age | Gender | MMSE | Disease Duration (y) |
Laterality in Atrophy* |
[11C]PiB |
|---|---|---|---|---|---|---|
| 1 | 85 | M | 20 | 2 | L | (-) |
| 2 | 82 | F | 25 | 6 | L | (-) |
| 3 | 82 | M | 22 | 6 | L | (-) |
| 4 | 87 | M | 14 | 11 | L | (-) |
| 5 | 81 | M | 18 | 5 | (-) | (-) |
| 6 | 83 | F | 15 | 1 | (-) | (-) |
| 7 | 85 | M | 28 | 7 | (-) | (-) |
| - | ||||||
| HC (n=12) | 71.8±8.7 | M/F=7/5 | 28.8±1.3 | n.a. | (-) | (-) |
Compared with the pathological diagnosis, the specificity of these clinical criteria ranges from 44.3% to 70.8% [8-10]. Novel fluid and imaging biomarkers to detect amyloid β (Aβ) and tau are known to increase the accuracy of the diagnosis of dementia [11-14]. We previously reported that among cases clinically diagnosed with AD, the number of Aβ-negative cases increases with age [15]. These Aβ-negative cases are not included in AD pathologically [16] nor by the recent clinical definition of AD used by the National Institute on Aging and Alzheimer’s Association research framework using biomarkers [8, 17, 18].
The classical amyloid hypothesis of AD suggests that oligomer or accumulated Aβ is the first and common pathway to induce neurodegeneration [19-21]. Tau accumulation and neuronal dysfunction may follow such Aβ accumulation [22, 23]. However, this sequence was mainly confirmed in hereditary AD [24, 25] and may not be applied to sporadic AD. Indeed, we previously reported that age-related accumulation of tau may precede robust Aβ accumulation, leading to AD [26]. Furthermore, the failure of many clinical trials targeting Aβ indicates that Aβ may not be the primary cause [27-30].
Neuropathologically, cerebrovascular diseases, primary Age-Related Tauopathy (PART), and α-synucleinopathy are the leading causes of non-amyloid dementia [6, 31-33]. Among these, age-related tau accumulation without Aβ in the medial temporal lobe, basal forebrain, and brainstem is postulated to represent PART [34-37]. The concept of PART covers cases with normal cognitive function to those with profound cognitive impairment. The latter cases are reported to include Argyrophilic Grain Disease (AGD) [38-41] and Neurofibrillary Tangle-Predominant Dementia (NFTPD) [42-45].
To explore the contribution of tau to Amyloid-Negative Dementia in the Elderly (ANDE), we studied tau accumulation with a radiopharmaceutically significant tracer, 2-[(1E, 3E)-4-[6-[11C] methylamino] pyridin-3-yl]buta-1,3-dien-1-yl]-1,3-benzothiazol-6-ol ([11C]PBB3), for Positron Emission Tomography (PET) [46-49].
2. MATERIALS AND METHOD
2.1. Ethics
Written informed consent was obtained from all participants or from close family when the participants were cognitively impaired. The present study was approved by the Institutional Research Ethics Committee of Osaka City University Graduate School of Medicine (IRB# 3009).
2.2. Healthy Controls
Subjectively Healthy Controls (HC) without a history of brain disorders were openly recruited as candidates. Detailed medical histories were obtained through interviews conducted by a board-certified neurologist. Thorough neurological examinations, including general cognitive function tests, were performed by the same neurologist. The Mini-Mental State Examination (MMSE), revised Hasegawa Dementia Scale (HDS-R) [50], and Rivermead Behavioral Memory Test (RBMT) [51] were conducted by qualified clinical psychologists (MA, NK). Blood samples were collected to evaluate general physical conditions. Magnetic Resonance Images (MRIs) were also evaluated.
Exclusion criteria were as follows: 1) history of any brain diseases, history of brain surgery, or head trauma requiring hospitalization; 2) high risks for cerebrovascular diseases, including poorly controlled diabetes, dyslipidemia, and high blood pressure; 3) any neurological findings suggesting brain disorders; 4) cognitive test (MMSE, HDS-R, and RBMT) indicating border-zone or worse level; and 5) MRI lesions including asymptomatic lacunas, severe white matter lesion, many cerebral microbleeds and atrophy beyond average for their age. 6) Amyloid PET imaging was employed for the exclusion of enrollment. No other biomarkers were examined in the HC group.
2.3. ANDE
Seven cases with slowly progressive dementia who were older than 80 years and were negative for Aβ were evaluated (age 83.5±2.1 years, 5 male, 2 female; (Table 1). The MMSE score was 19.9±4.5 and the disease duration was 5.4±3.3 years.
In one case (case 1), an autopsy was obtained 2 years after the PET examination. The section of the brain was examined with Gallyas Braak silver 4 and immunostained using the following antibodies; phosphorylated tau (p-tau; AT-8, 1:100, monoclonal; Innogenetics, Ghent, Belgium), Aβ (Aβ11-28, monoclonal; IBL, Maebashi, Gunma, Japan), phosphorylated α-synuclein (p Syn#64, polyclonal, 1:20,000; Wako, Osaka, Japan), and phosphorylated TDP-43 (p-TDP-43) (pSer409/410, monoclonal, 1:10,000; Cosmo Bio, Tokyo, Japan).
Among cases without an autopsy, three cases showed strong laterality in hippocampal atrophy on MRI, and AGD was clinically suspected. The left side showed greater atrophy in all cases. The other 3 cases had no significant laterality in hippocampal atrophy (probable NFTPD).
2.4. PET Data Acquisition
[11C]PBB3 and [11C]Pittsburgh compound-B (2-[4-([11C] methylamino) phenyl]-1,3-benzothiazol-6-ol, [11C]PiB) were produced based on the previously reported method [46, 52, 53]. [11C]PBB3-and [11C]PiB-PET images were acquired with a Siemens Biograph16 scanner (Siemens/CTI, Knoxville, TN, USA) and with an Eminence-B PET scanner (Shimadzu Co., Kyoto, Japan), respectively. For tau imaging, [11C]PBB3 in the range of 370 MBq (Body Weight ≤50kg) to 555 MBq (Body Weight ≥70kg) was intravenously injected in a dimly lit room to avoid photoracemization. A 60-minute PET scan was performed in the list mode. The acquired data were sorted into dynamic data of 6×10 s, 3×20 s, 6×60 s, 4×180 s, 8×300 s frames. For Aβ imaging, each subject received 400 to 500 MBq of [11C]PiB intravenously over 1 minute. After the injection, static scan image acquisition was performed for 50 to 70 minutes. PET images for [11C]PBB3 and [11C]PiB were reconstructed by filtered back projection using a 4-mm Full Width at Half Maximum (FWHM) Hanning filter and a 5-mm FWHM Gaussian filter with attenuation and scatter correction, respectively.
2.5. MRI Acquisition
MRI data were obtained with a 1.5 or 3-Tesla magnetic resonance scanner (MAGNETOM Avanto, Siemens Healthcare, Erlangen, Germany or Ingenia, Philips Healthcare, Best, The Netherlands). Three-dimensional volumetric acquisition of a T1- weighted gradient echo sequence (repetition time range/echo time range, 6.5 ms/3.2 ms; field of view [frequency × phase], 240 mm×240 mm; matrix, 256×256; contiguous axial slices of 1.5 mm thickness) was performed.
2.6. Image Processing
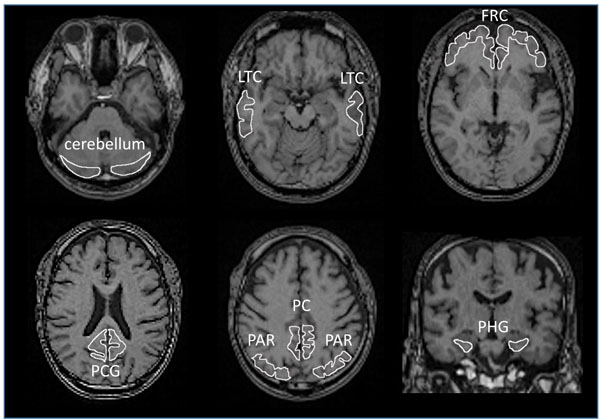
All images were processed using PMOD software version 3.7 (PMOD Technologies Ltd., Zurich, Switzerland) [54-56] and Statistical Parametric Mapping software (SPM12, Wellcome Department of Cognitive Neurology, London, UK), operating on the MATLAB software environment (version R2016b; MathWorks, Natick, MA, USA) [57]. Acquired [11C]PBB3 and [11C]PiB images were transformed into the standard brain using PMOD, and then, SUVR images were generated with the cerebellar cortex as a reference region using the frame-summation of dynamic image for 30 to 50 minutes after [11C]PBB3 injection and for 50 to 70 minutes after [11C]PiB injection. SUVR level of each VOI was calculated in manually set ROI as shown in Fig. (1). To facilitate the visual presentation of SUVR images, [11C]PBB3 accumulation outside of the masked brain was eliminated. Mask images of the brain, set on MRI images of each case, were transformed to match the standard brain.
Each T1-weight MRI was segmented and anatomically normalized to MNI152 standard space (Montreal Neurological Institute, Montreal, QC, Canada) using SPM12 and Diffeomorphic Anatomical Registration Through Exponentiated Lie Algebra (DARTEL) [58]. The [11C]PBB3-SUVR images were co-registered to individual T1-weighted MRI using PMOD and then normalized to MNI space using the same parameters as those for MRI (flow fields method) with smoothing at 8-mm full-width-at-half-maximum.
2.7. Criteria for Aβ Accumulation
Aβ accumulation was visually evaluated based on the Japanese Alzheimer’s Disease Neuroimaging Initiative (J-ADNI) Visual Criteria for [11C]PiB-PET (J- ADNI _ PETQC _ Ver1.1) modified from ADNI PET core criteria [59]. Four regions selected for the assessment were the precuneus-posterior cingulate gyrus, frontal lobe, lateral temporal lobe, and lateral parietal lobe. When the accumulation in one of the above 4 cerebral cortices was higher than that in the white matter just below the cortex, the case was determined as positive. When none of the 4 cortices had higher accumulation than the white matter, the case was confirmed as negative.
2.8. Voxel-Wise Analysis of [11C]PBB3
Voxel-wise comparison of [11C]PBB3 accumulation data was performed between the ANDE and HC group by 2-sample t-test using SPM12 [60]. In addition, [11C]PBB3 data from individual ANDE case was compared with the HC group by Jack-knife analysis using SPM12 [61]. In both analyses, the threshold for significance was set at P<0.05 Family-Wise Error (FWE)-corrected at a cluster-level following P<0.01 uncorrected at the voxel level.
2.9. Statistical Analysis of Regional SUVR
In each regional SUVR, age correction was performed using by the linear regression set for the data of HC. Details are described elsewhere [26]. In short, the age-dependent linear increase in regional SUVR was found in all ROI’s in the HC group and the linear regression curve was calculated to the data in each ROI. Age correction for SUVR in each case was made based on the linear curve. Group comparison was made between HC and AGD or between HC and NFTPD. To evaluate the effect of atrophy on apparent [11C]PBB3 accumulation in AGD, each side was separately analyzed. Accumulation of [11C]PBB3 on the side of less atrophy was compared with that of more atrophy by paired t-test.
3. RESULTS
We found that there was a very high burden of tau in the lateral temporal cortex in ANDE. Scattered neurofibrillary tangles were confirmed in this region in an autopsied case. In addition, diffuse PBB3 accumulation was found in the bilateral parietal lobes, precuneus, and parahippocampal gyrus. No corresponding tau pathology was confirmed in the autopsied case in these regions, suggesting possible undetermined binding pathology in ANDE.
3.1. Demographical Data and MRI Findings
Table 1 shows the demographical data of ANDE and HC. Cases of ANDE were older than HC. The method for age correction in the image analysis was stated above. Four cases out of 7 in ANDE had an MMSE score less than or equal to 20 points. Four cases of clinical AGD had longer disease duration, and higher MMSE score than NFTPD cases, reflecting the general characteristic of these diseases. Due to the small sampling size, a statistical comparison was not made.
3.2. Tau Accumulation in ANDE
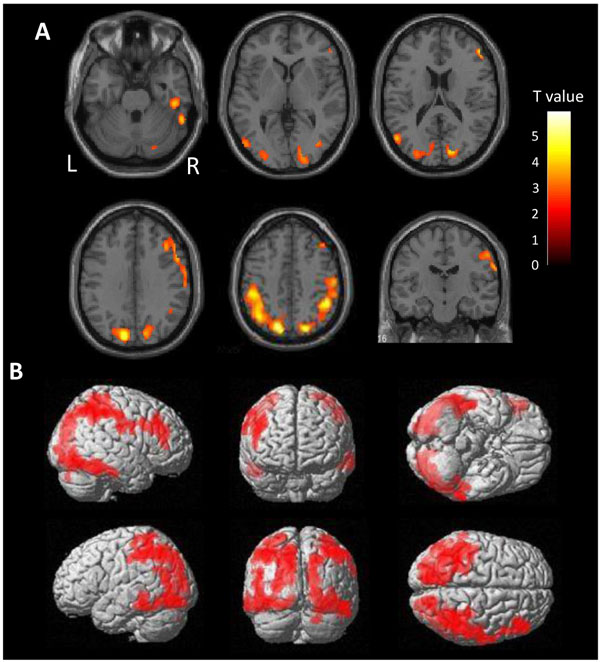
There is a very high burden of tau in the diffuse area of the bilateral parietal lobes in ANDE by voxel-wise comparison with HC (Fig. 2). Mild accumulation was found in the frontal lobes and lateral temporal lobes on both sides. Accumulation in the cortices neighboring the sagittal sinus and the transverse sinus may reflect spillover of nonspecific accumulation in the sinuses. Cortical accumulation was observed to be centered on the gyrus, not on the sulcus, excluding the possibility of vascular spillover on the cortex.
3.3. Regional SUVR of [11C]PBB3 in ANDE and HC
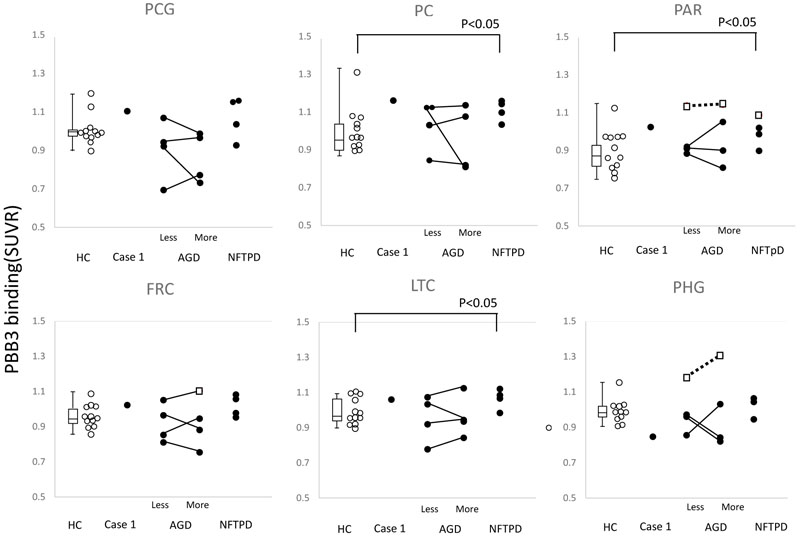
The SUVR of AGD and NFTPD was compared with that in HC. Significantly high accumulation was found in the parietal cortex, precuneus and the lateral temporal cortex of NFTPD (P<0.05) (Fig. 3).
To evaluate the effect of atrophy on apparent [11C]PBB3 accumulation, each side was analyzed separately in cases with AGD. In most cases, a great difference in [11C]PBB3 accumulation was seen between the sides, but no correlation was found between the side of atrophy and level of tau accumulation (Fig. 3).
In addition, the level of individual SUVR was compared with the distribution of tau accumulation in HC. The open square in Fig. (3) indicates significantly high accumulation in the parietal lobe, frontal lobe, and parahippocampal gyrus.
No correlation was found between MMSE and SUVR in AGD as well as in NFTPD.
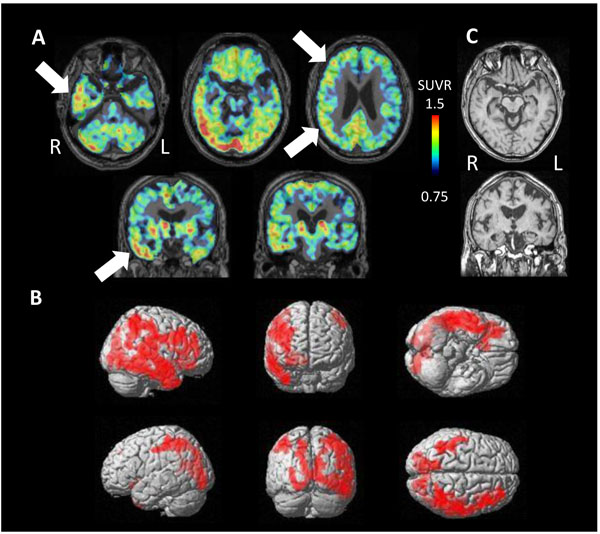
3.4. Autopsy Case
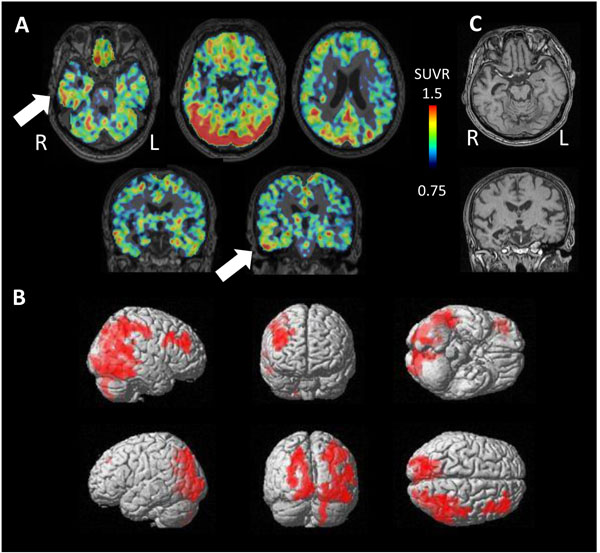
PET study revealed tau accumulation at the right lateral temporal lobe, frontal lobe and, parietal lobe (Fig. 4A,B). Cortical atrophy was predominant on the left side (Fig. 4C), where an apparent accumulation of tau was less severe.
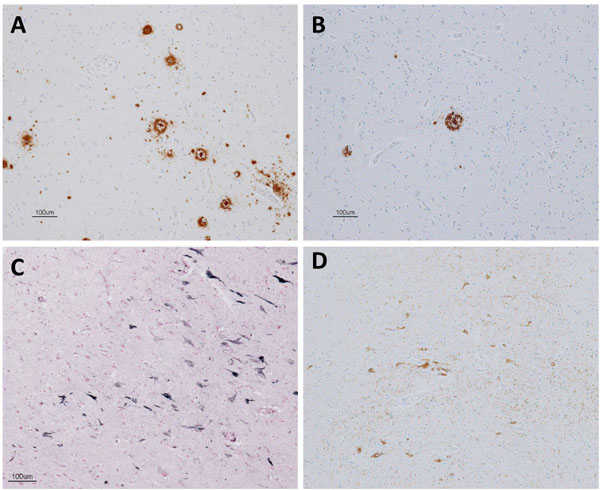
Pathologically, a very limited number of senile plaques immunopositive for Aβ11-28 were found in the temporal lobe (Fig. 5A), occipital lobe and parietal lobe (Fig. 5B; Braak amyloid stage A). Neurofibrillary tangles were scattered in the parahippocampal gyrus (Gallyas-Braak silver staining, Fig. 5C; Braak neurofibrillary stage II) and were immunopositive for AT8 antibody (Fig. 5D) [16, 62, 63]. Neither argyrophilic grain, Lewy body, nor TDP-43 positive structure was found. In conclusion, pathological diagnosis was mild ageing change.
3.5. Subtypes of ANDE
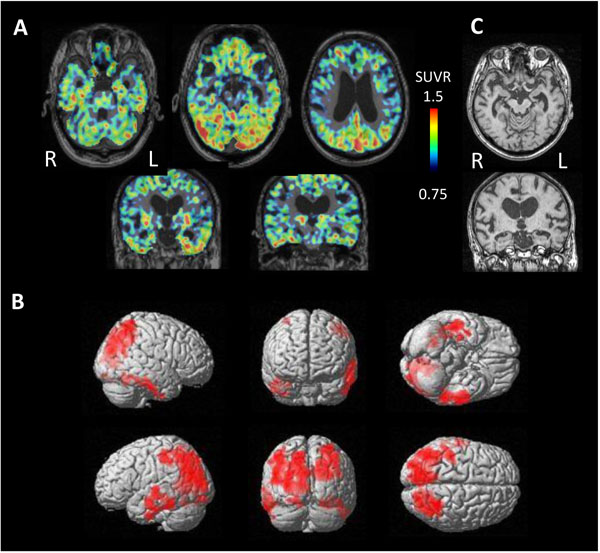
Four cases were diagnosed as having AGD based on clinical course and neuroimaging findings. These cases showed significantly high tau accumulation on either side of the temporal lobe (Fig. 6). The side of higher tau accumulation was on the atrophied side in 3 of 4 cases, suggesting the dominantly affected side.
In contrast, 3 cases of NFTPD revealed tau accumulation on the bilateral temporal lobes and parietal lobes (Fig. 7). MRI showed mild atrophy on the bilateral parahippocampal gyrus.
4. DISCUSSION
4.1. General Comments
To explore the mechanism of dementia often found in the ANDE, we studied the possible involvement of tau with PET. Compared to the age-corrected HC, we found that tau accumulation is significantly high in the parietal lobe of cases with dementia. Together with the lesion at the precuneus and temporal lobe in some cases, we speculate that the tau accumulation detected with [11C]PBB3-PET induces dementia. We previously reported that these ANDEs result in milder dementia and progress more slowly than AD [64]. Furthermore, amyloid targeting therapy must be ineffective in amyloid negative dementia. Therefore, distinguishing between these 2 pathophysiologies is critical.
The concept of SNAP (Suspected Non-Alzheimer Pathology) covers young as well as old cases [31, 33, 65]. It also includes cases with normal cognitive function as well as those with mild to severe dementia. By definition, ANDE in the present study is included in SNAP.
4.2. Pathological Mechanisms for Tau Accumulation
An autopsy was obtained in only 1 case, in which neurofibrillary tangles were observed at the parahippocampal gyrus. [11C]PBB3-PET also revealed tau accumulation in the same lesion. Three cases of clinical NFTPD showed significant [11C]PBB3 accumulation at lateral temporal lobe. These are the typical distributions of neurofibrillary tangle in NFTPD, suggesting the possibility that [11C]PBB3 detects tau in the neurofibrillary tangle in these cases.
Four cases of clinical AGD showed inconsistent results of [11C]PBB3 accumulation, some high and the others low. The side of atrophy does not coincide with that of high [11C]PBB3. High accumulation of AGD may induce cerebral atrophy, but further atrophy may reduce apparent tau accumulation, even below the level of the opposite side. Partial volume effect might enhance this apparent reduction on [11C]PBB3 images. Similar effects of cortical atrophy on apparently less tau accumulation may explain the opposing laterality in the autopsied case.
Parietal accumulation of PBB3 was significant in ANDE. Unexpectedly, no tau pathology was found in the parietal lobe of the autopsied case. In the present study, the average age of ANDE was higher than that of HC. Although the age-dependent increase in PBB3 binding was corrected with the linear regression curve, aging effect may be stronger than expected in this region. Another possibility is that undetermined pathology with PBB3 binding in ANDE might have developed in the parietal cortex. Further pathological study is warranted.
4.3. Tau Accumulation in Aging, Amyloid-Negative Dementia and AD
We previously reported that tau accumulates with age in the region related to AD [26]. This is in good accordance with the previous reports by Jagust WJ et al. regarding [18F]-AV-1451 [65-67]. In the present study, we additionally found a further accumulation of tau in these regions, especially in the parietal lobe, in ANDE patients for the first time. Recently, age-related tau accumulation without Aβ in the medial temporal lobe, basal forebrain and, the brainstem is postulated to represent Primary Age-Related Tauopathy (PART) [34, 35, 68]. The concept of PART covers the case with normal cognitive function to that with profound cognitive impairment. [11C]PBB3 may detect this whole spectrum of PART.
In contrast, we previously reported that amyloid and tau accumulation begins in Mild Cognitive Impairment (MCI due to AD) and reaches the high level in AD [26]. Amyloid and tau may interact with each other to develop further lesions in AD [69-71].
4.4. Characteristics of [11C]PBB3
One of the drawbacks of [11C]PBB3 is that it accumulates at cerebral sinuses including the superior sagittal sinus and transverse sinus [72-74]. The extent of accumulation varies between cases. Low accumulation can be eliminated by masking the accumulation at the sinuses, although high accumulation can affect the apparent measurement of the cerebral cortex. In the present study, high accumulation neighboring superior sagittal sinus and transverse sinus in the occipital lobe was considered an artifact.
The limited dynamic range of [11C]PBB3 to detect tau accumulation also may overlook smaller lesion or milder accumulation [48]. In the present study, tau accumulation was smaller and a milder lesion at parahippocampal gyrus and lateral temporal cortex may be underestimated in the study of [11C]PBB3. Possible overestimation of aging effect on tau accumulation at these lesions may additionally reduce the significance [26]. Further development of more sensitive tracer is warranted [75].
4.5. Study Limitation
Pathology was confirmed only in 1 case, in which neurofibrillary tangles were identified abundantly in the parahippocampal gyrus but not so many in the parietal lobe. There might be some gap between [11C]PBB3 accumulation and pathological changes. Although the distribution of tau by [11C]PBB3 in cases with NFTPD in the present study is consistent with the distribution of lesion in the previous pathological reports, further pathological confirmation is warranted. Subjects were chosen from patients with clinical NFTPD and AGD at an outpatient clinic. A greater number of subjects with ANDE in general should be studied to draw definite conclusions.
CONCLUSION
A relatively high burden of tau deposition was commonly observed in the lateral temporal cortex of ANDE, which may explain dementia in these subjects. [11C]PBB3 may be useful in detecting tauopathy in these cases.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The present study was approved by the Institutional Research Ethics Committee of Osaka City University Graduate School of Medicine (IRB# 3009).
HUMAN AND ANIMAL RIGHTS
No animals were used in this research. All human research procedures were followed in accordance with the ethical standards of the committee responsible for human experimentation (institutional and national), and with the Helsinki Declaration of 1975, as revised in 2013.
CONSENT FOR PUBLICATION
Written informed consent was obtained from all participants or from close family when the participants were cognitively impaired.
FUNDING
The present study was financially supported by the NIRS/QST grant (Japan Agency for Medical Research and Development 16768966), in which Osaka City University was involved as a collaborating facility.
CONFLICT OF INTEREST
The author declares no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
The authors thank Ms. M. Ando and Ms. N. Kotani (Osaka City University) for psychological tests.

