All published articles of this journal are available on ScienceDirect.

Interactions of Human Endothelial and Multipotent Mesenchymal Stem Cells in Cocultures
Authors Info & Affiliations
Abstract
Current strategies for tissue engineering of bone rely on the implantation of scaffolds, colonized with human mesenchymal stem cells (hMSC), into a recipient. A major limitation is the lack of blood vessels. One approach to enhance the scaffold vascularisation is to supply the scaffolds with endothelial cells (EC).
The main goal of this study was to establish a coculture system of hMSC and EC for the purposes of bone tissue engineering. Therefore, the cell behaviour, proliferation and differentiation capacity in various cell culture media as well as cell interactions in the cocultures were evaluated.
The differentiation capacity of hMSC along osteogenic, chondrogenic, and adipogenic lineage was impaired in EC medium while in a mixed EC and hMSC media, hMSC maintained osteogenic differentiation. In order to identify and trace EC in the cocultures, EC were transduced with eGFP. Using time-lapse imaging, we observed that hMSC and EC actively migrated towards cells of their own type and formed separate clusters in long term cocultures. The scarcity of hMSC and EC contacts in the cocultures suggest the influence of growth factor-mediated cell interactions and points to the necessity of further optimization of the coculture conditions.
INTRODUCTION
In the field of bone tissue engineering, human mesenchymal stem cells (hMSC) are commonly used as osteoprogenitor cells for colonization of scaffolds to be implanted in the injured tissue [1-3]. Adult hMSC are defined mainly by their capacity of differentiating along multiple lineages into end-stage mesenchymal cell phenotypes [4-7]. These include bone/osteoblasts [8,9], cartilage/chondrocytes [10,11], fat/adipocytes [12,13] as well as a variety of other connective tissues such as muscle [14] or stroma [15]. In addition, the hMSC secrete a variety of cytokines and growth factors that have both paracrine and autocrine activities [16-19].
Osteogenic and endothelial cells (EC) have been demonstrated to closely associate in the process of bone regeneration and to produce growth factors that affect the growth and differentiation of the reciprocal cell type [20,21]. This implies that presence of EC on the implanted scaffolds may help to achieve an optimal colonization of the tissue constructs. The hMSC cultured in direct contact with EC manifested an increased osteogenic differentiation in vitro [22]. In vivo, cotransplantation of EC with hMSC resulted in more bone formation than did the transplantation of hMSC alone [23]. In contrast, only few hMSC survived when implanted on scaffolds into athymic nude mice [24].
The combination of EC and osteoprogenitor cells is expected to not only accelerate the vascularization essential for delivery of nutrients to the developing bone but also to be beneficial for the growth and differentiation of both cell types. Osteoprogenitor cells are known to secrete VEGF and could therefore enhance the proliferation and differentiation of EC [21,25], on their turn, can enhance the proliferation and differentiation of osteoprogenitor cells by the secretion of osteogenic growth factors [26,27].
The goal of this study was to examine the interactions of hMSC and EC in cocultures. We cultured hMSC from three different donors with EC and analyzed the capacity of hMSC to proliferate and to differentiate in growth media supporting the growth of both hMSC and EC. As endothelial cell model, we used the human endothelial cell line AS-M.5 which has demonstrated to manifest the characteristics of freshly isolated endothelial cells [28,29]. Finally, we observed both short and long term development of cocultures of hMSC and EC as well as the interactions of the two cell types.
MATHERIALS AND METHODS
Cell Culture
The hMSC from 3 different caucasian donors of 18 (donor I), 19 (donor II), and 25 (donor III) years of age (Lonza, Basel, Switzerland) were used and expanded in Mesenchymal Stem Cell Growth Medium (Lonza) for one week before being used in the experiments. Endothelial cell line AS-M.5 cells [28,29] were propagated in Endothelial Cell Growth Medium MV (PromoCell, Heidelberg, Germany). This low serum (5%) medium contains no additional growth factor supplements like VEGF of EGF-2 that may influence the dynamics between cell populations. The cells were plated at a density of 2000 cells/cm2. The 3 test media were M1: cell culture medium developed for expansion of hMSC, supplemented with FGF-2 to enhance their mitogenic potential [30] containing D-MEM high Glucose with L-Glutamine and Sodium Pyruvat, (PAA Laboratories, Pasching, Austria), 10 % fetal bovine serum (FBS), Penicilin- Streptomycin, (Sigma Aldrich, St. Louis, USA), and 5 ng/ml Fibroblast Growth Factor 2 (FGF-2, RELIATech, Braunschweig, Germany), M2: M1 and M3 mixed in a ratio of 1:1, and M3: Endothelial Cell Growth Medium MV (PromoCell). Medium M2 was designed for the propagation of the two cell types we studied in cocultures in order to maintain enough factors in the medium to sustain the viability of both cell types without affecting/ inducing their differentiation. Cells were subcultured by trypsinization, and reseeded at a density of 2000 cells/cm2 after 7 and 14 days of culture. Prior testing the effects of cell culture medium, cells were kept in each particular medium for 21 days, as indicated in Fig. (1).
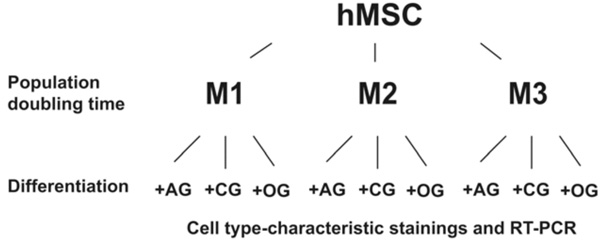
Flowchart of cultivation protocols and assays. The population doubling times of hMSC were determined in media M1 (D-MEM high Glucose with 10 % FBS and Penicilin- Streptomycin), M2 (M1 and M3 mixed in a ratio of 1:1), and M3 (Endothelial Cell Growth Medium MV). The differentiation capacity of hMSC was analyzed by culturing the cells in media promoting the differentiation along different lineages, followed by cell type specific staining as well as by RT-PCR. AG – adipogenic supplements; CG – chondrogenic supplements; OG – osteogenic supplements.
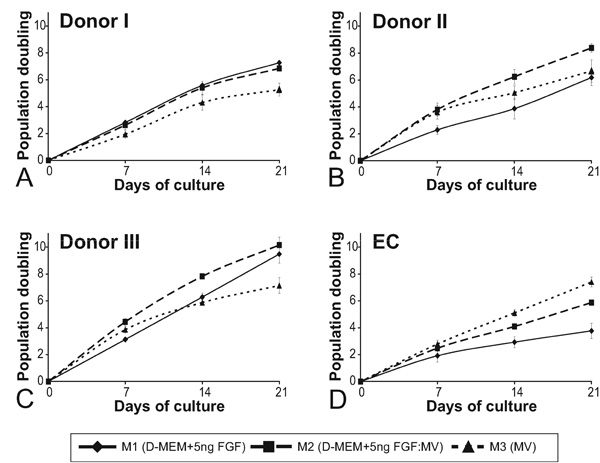
The proliferation rate of hMSC is donor dependent. Cumulative population doublings of hMSC from 3 different donors (A, B, C) and AS-M.5 (D) in cultures supplemented with M1 (diamonds, full line), M3 (triangles, dashed), and M2 (squares, dashed line) in the course of 21 days of cultivation. Indicated are means +/- SEM.
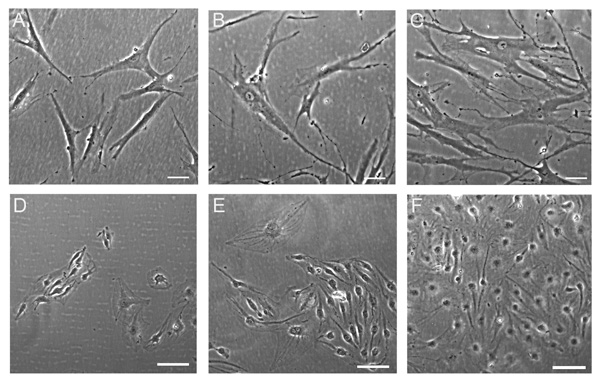
Cell culture media affect the morphology of hMSC. The hMSC (A, B, C) and AS-M.5 (D, E, F) were grown in medium M1 (A, D), medium M2 (B, E), and medium M3 (C, F) for 18 days. Phase contrast, bar represents 100 µm.

Growth media affect the differentiation potential of hMSC in vitro. The hMSC were grown in M1 (A, D, G), M2 (B, E, H), and M3 (C, F, J) for 21 days, and subsequently subjected to differentiation. The osteogenic differentiation was detected by means of von Kossa staining (A, B, C), adipogenic differentiation by means of oil red O staining (D, E, F), and chondrogenic differentiation by means of collagen II staining (G, H, J). Bar represent 200 µm (B), 100 µm (A, C, G insert), 50 µm (D, E, F), and 300 µm (G, H, J).
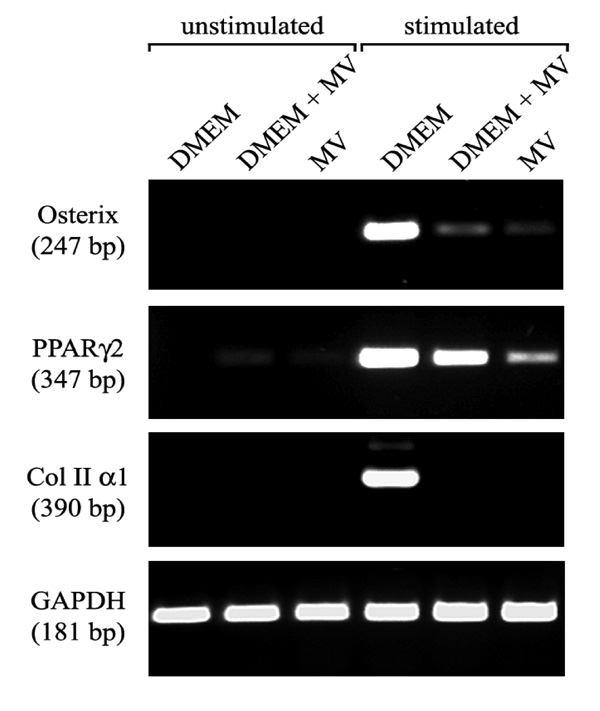
Expression of characteristic differentiation marker is dependent on the growth medium. RT-PCR analysis of gene expression of hMSC cultured in the three types of growth medium (M1, M2, M3), and subsequently in medium inductive for osteogenic, adipogenic, and chondrogenic differentiation. The expression of marker characteristic for each lineage was assayed by RT-PCR. Osterix transcription factor for osteogenic, PPARγ2 transcription factor for adipogenic and collagen type II for chondrogenic differentiation. RNA integrity was assessed by GAPDH amplification.
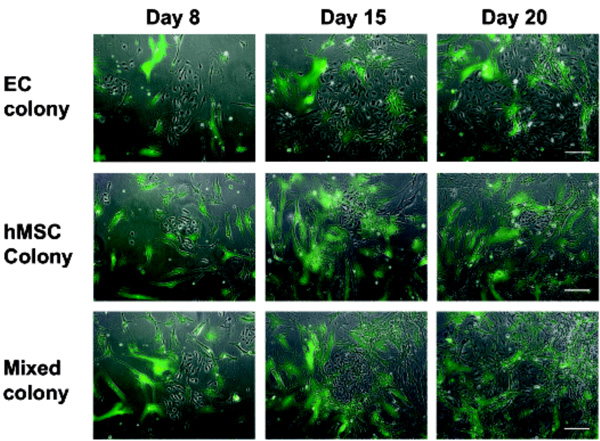
The hMSC and EC form distinct colonies in cocultures. Mixtures of EGFP-transduced hMSC and EC were propagated in the coculture medium for 21 days. Shown are typical clusters of endothelial cells: loose colonies associated with few hMSC (colony 1), small dense clusters amidst sparsely distributed hMSC (colony 2), and compact colonies covered with few hMSC (colony 3). Bar represents 100 µm.
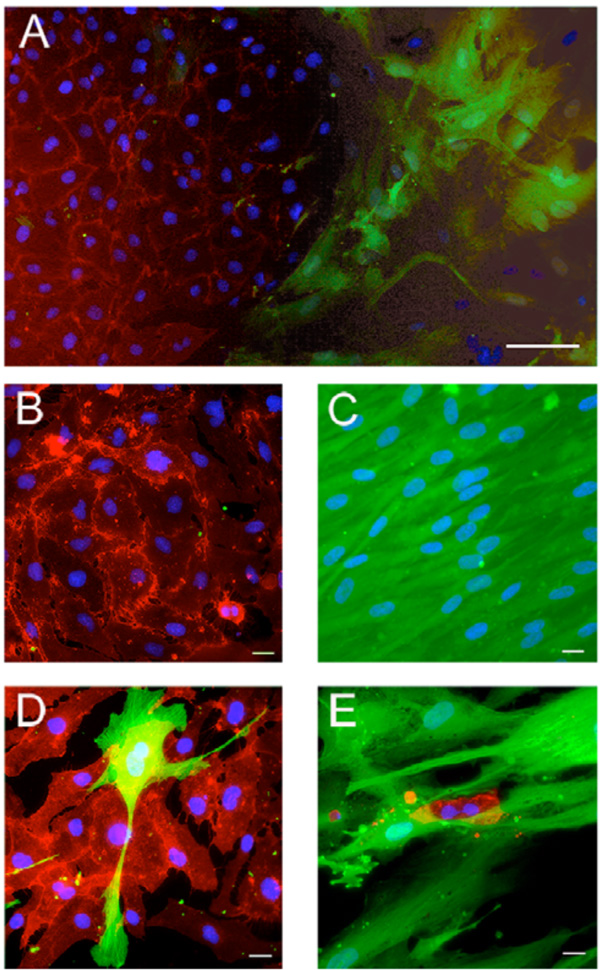
Appearance of the cocultures of hMSC and EC. Mixtures of EGFP-transduced hMSC and AS-M.5 were cultured in M2 medium for 21 days. CD31-positive endothelial cells were detected by immunofluorescence. In cocultures (A), both cell typed developed distinct clusters containing either endothelial cells (B) or hMSC (C). Close cell contact of hMSC and EC were observed only sporadically (D, E). Bar represent 100µm (A), 20 µm (B-E).

Both hMSC and EC actively migrate to build clusters. The behaviour of EGFP-transduced hMSC (green fluorescence) and EC in the cocultures was evaluated 30h after plating. The cell migration was observed on time-lapse pictures taken every 5 min for 12h. The arrow indicates a single hMSC migrating towards another hMSC that is marked with a star. Bar represents 100 µm.
Cocultures
For long-term experiments, cocultures were initiated by plating cell suspensions containing hMSC-EGFP und AS-M.5 in equal proportion at a density of 500 cells/cm2 in 25 cm2 tissue culture flasks in M2 medium. The development of the cultures was followed over 3 weeks, medium was changed twice weekly. In short-term experiments, hMSC-EGFP and AS-M.5 were plated at a density of 3000 cells/cm² on 35 mm cell culture dishes (BD Falcon, San Jose, USA) and cultured for 6, 30, and 54 h. The time-lapse pictures were taken at interval of 5 min for 12h using digital camera AxioCam MRm mounted on Axiovert S100 (Carl Zeiss).
Induction of Differentiation and Evaluation of Differentiation Potential of hMSC
Differentiation was induced by propagating hMSC in the culture media containing differentiation supplements. Briefly, 5 x 103 cells/cm2 were seeded in the maintenance medium (M1, M2 or M3), and cultured till 80 % confluency.
Osteogenic differentiation of hMSCs was assessed using a well-established assay [7]. Stimulation with osteogenic supplements 100 nM dexamethasone, 10 mM β – glycerophosphate, and 50 µM L-ascorbic acid-2-phosphate (all Sigma Aldrich) started after the cells reached confluence of about 80 %. Medium was changed twice weekly.
Chondrogenic differentiation was induced in high density pellet cell cultures as previously described [7]. Briefly, 2.5 x 105 cells were centrifuged (500g, 5 min, RT), the cell pellets were cultured in 1 ml of medium without FBS but containing chondrogenic supplements: 100 nM dexamethasone, 1mM pyruvate, 195 µM L-ascorbic acid-2-phosphate, 350 µM L-proline, 1,25 % (v/v) insulin-transferrin- selenious acid mix (ITS, 100x), 5,35 µg/ml linolic acid, 1,25 mg/ml bovine serum albumine (BSA) (Sigma Aldrich). Finally, TGF- β3 (R&D Systems, Minneapolis, USA) was added to a final concentration of 10 ng/ml, and then incubated in a 95% humidified atmosphere, 5% CO2 at 37°C for 5 weeks. The medium was changed 2 – 3 times a week. Chondrogenic differentiation was assayed after 5 weeks of stimulation.
Adipogenic differentiation was induced according to a published protocol [7]. Stimulation with adipogenic supplements (1 µM dexamethasone, 0.2 mM indomethacin, 0.1 mg/ml insulin, 1 mM 3-isobutyl-1-methylxanthin (IBMX) (Sigma Aldrich) started after the cells reached full confluence. After five days of growth in the adipogenic stimulation, medium the cultures were set in the maintenance medium for two days, and finally in the stimulation medium again. The medium was changed three times a week.
Cells cultured in parallel in the medium lacking differentiation supplements were used as negative controls.
Detection of Differentiated Cells
Differentiated cells of osteogenic lineage were detected after 21 days of stimulation by von Kossa staining using a standard protocol.
To evaluate chondrogenesis in the aggregates, the cultures were harvested and subsequently frozen in Jung tissue freezing medium (Leica Instruments, Wetzlar, Germany). Twelve µm sections were cut with a cryostat vacutome HM 500 OM (Microm, Walldorf, Germany). Type II collagen was detected using mouse monoclonal antibody CIIC1 (DSHB, University of Iowa, Iowa City, USA), biotinylated anti-mouse secondary antibody (Vectastain Elite ABC Kit Mouse IgG) in combination with the 3,3´-diaminobenzidine DAB Substrate Kit (both Vector Laboratories, Burlingame, USA) according to the instructions of the manufacturer. The sections were counterstained in Mayer´s hemalaun.
Adipogenic cells were detected by oil red O staining. The cultures were fixed with 3,7 % formaldehyde (Merck, Darmstadt, Germany) for 2 minutes at -20 °C, washed with chilled 50 % ethanol, stained by incubation with 0,2 % (w/v) oil red O solution (Sigma Aldrich), and subsequently photographed with a Sony digital camera (DSC-S75, Sony, Tokyo, Japan) attached to an Axiovert S100 microscope (Carl Zeiss, Jena, Germany).
Immunofluorescence Analysis
Cells (1 x 104 cells/well) were cultured on 8-well culture slides (BD Biosciences, San Jose, USA) for 24h, fixed with 3.7% formaldehyde in cytoskeleton stabilizing buffer (CS: 0.1 M PIPES, 1 mM EGTA, 4% [w/v] polyethylene glycol 8000 (all Sigma-Aldrich), 137 mM NaCl, 5mM KCl, 1.1mM Na2 H2 PO4, 0.4 mM K2 H2 PO4, 5.5 mM glucose, 5 mM PIPES, 1 mM MgCl2, 2 mM EGTA (pH 6.9), [31], and exposed to the primary antibodies M0823 (DAKO, Glostrup Denmark, Germany) and 5561 (BD Biosciences) to detect the characteristic endothelial proteins CD31 and CD144, respectively. Secondary antibody was Alexa Fluor 488 anti-mouse IgG (Molecular Probes, Leiden, The Netherlands). The staining was evaluated using fluorescence microscope Axioscope 2 and digital camera AxioCam MRm (Carl Zeiss).
RNA Isolation and RT-Polymerase Chain Reaction (PCR) Analyses
Total cellular RNA was isolated using RNeasy mini kit (Qiagen, Hilden, Germany) and reverse-transcribed using the First-Strand cDNA Synthesis Kit (Invitrogen, Carlsbad, USA) according to the instructions of the manufacturer. The expression of the transcripts encoding osterix, PPAR γ2, and human collagen II was assessed with specific primer pairs [32] and using PCR mix (Invitrogen). Amplifications were performed in a PTC-200 cycler (MJ Research, Walthem, USA) for 25 - 35 cycles, (typically: 95°C for 30 s, 50-55°C for 30 s, 72°C for 60 s after an initial denaturation at 95°C of 3 minutes, and followed by the final extension at 72°C for 10 minutes. To control the integrity of cDNA, the house-keeping glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was amplified in parallel reactions [33]. PCR products were analysed on 1.8% agarose gel and visualised by ethidium bromide staining.
Lentiviral Vector Constructs, Lentivirus Production, and Transduction of Target Cells
The cDNA encoding the enhanced green fluorescent protein (EGFP) from the plasmid pEGFP-N3 (Takara Bio Europe /Clontech, Saint-Germain-en-Laye, France) was ligated into the lentiviral vector pLenti6V5-DEST (Invitrogen). Lentivirus was produced using the ViraPower lentiviral expression system (Invitrogen) according to the instructions of the manufacturer with minor adjustments [34]. Viral supernatant was used in a 1:2 dilution in normal growth media for transduction of 6 x 103 hMSCs / cm2. The transduced cells were selected with 5 µg/ml blasticidin S HCl (Invitrogen). The eGFP- transduced hMSCs were able to differentiate towards adipogenic, osteogenic, and chondrogenic lineages suggesting that they have fully retained their stem cell character [34].
RESULTS
Development of hMSC and EC Cultures in Various Growth Media
The effects of growth medium on the proliferation and differentiation capacity of hMSC and EC were examined as indicated in the flow chart (Fig. 1). Searching for a medium that support the growth of both hMSC and EC in cocultures, we evaluated the cell growth rate of hMSC in medium M1 (DMEM + FGF-5ng/ml), M2 (1: 1 mixture of M1 and M3), and M3 (endothelial cell growth medium MV).
Major differences in the proliferation rate of hMSC isolated from three different donors were observed in the three media tested. The cumulative population doubling of hMSC in the course of 21 days of cultivation have shown significant donor-dependent differences (Fig. 2). While hMSC of donor II and III (Fig. 2B, C) proliferated in M3 more rapidly than in M1 during the first two weeks, the growth of hMSC of donor I was in M3 much slower than in the M1. The hMSC of all 3 donors proliferated in M2 either as fast as in the M1 or faster. Similarly, virtually no proliferation of EC was observed in M1 but a slow proliferation of EC was measured in the M2. Even though the cultures of hMSC isolated from 3 different donors proliferated continuously in the M1 in the course of the observation period of 21 days, they differed substantially in the number of population doubling they ultimately reached. While the cultures of donor III reached population doubling of 10, the cultures of the remaining donors did not surpass 7 population doublings. The hMSC cultures isolated from different donors differed also in their capacity to proliferate in M3. While hMSC cultures from donor I and III ceased to proliferate exponentially in M3 after about 2 weeks in culture, the cultures from the donor II continued to proliferate in M3 at a similar rate as in the M1.
The hMSC cultures of all three donors tested showed similar cell morphology in response to the three growth media tested (Fig. 3). While a large proportion of hMSC grown in M1 manifested the typical fibroblast-like morphology (Fig. 3A), giant senescent cells were observed in the hMSC cultures propagated either in M2 (Fig. 3B) or in M3 (Fig. 3C). Endothelial AS.M-5 showed their typical morphology in M3 (Fig. 3F) whereas few morphologically aberrant cells were observed in M2 and M1 (Fig. 3D, E).
Differentiation Capacity of hMSC
In order to evaluate the effects of growth media on the differentiation potential of hMSC and to prove their stem cell character, we propagated the hMSC of the three donors in the three growth media for 21 days and subsequently subjected the cells to treatments known to induce osteogenic, chondrogenic, and adipogenic differentiation. Osteogenic differentiation could be induced in hMSC propagated in all three media tested, as shown by means of von Kossa staining (Fig. 4A, B, C). In addition, the expression of the osteogenic differentiation marker osterix in the cells that have been propagated in M1, M2 or M3 was demonstrated by RT-PCR (Fig. 5).
Chondrogenic differentiation could be induced only in the hMSC cultures propagated in M1. The expression of collagen II, the marker for chondrogenic differentiation, was detected both by immunohistological staining (Fig. 4G) and by RT-PCR (Fig. 5). In contrast, no expression of collagen II could be detected either by immunohistological staining (Fig. 4H, J) or by means of RT-PCR, in the hMCS propagated in either M2 or M3.
An accumulation of triglycerides in the cytoplasm after the induction of adipogenic differentiation was observed hMSC grown in all three media as demonstrated by oil red O staining (Fig. 4D, E, F). However the hMSC cultures growth in M2 and M3 were less dense than those grown in M1. Accordingly, less transcripts encoding peroxisome proliferator activated receptor γ (PPAR-γ) could be detected in the cultures grown in M2 and M3 (Fig. 5).
To ensure, that the endothelial supplements included both in M2 and M3, do not induce endothelial differentiation of hMSC, we stained hMSC propagated in these media for the characteristic endothelial markers CD31 (PECAM-1) and CD144 (VE-cadherin). No CD31 or CD144 expression could be detected. AS-M.5 propagated in M2 preserved the expression of these typical endothelial cell surface marker molecules (data not shown).
Cocultures of hMSC and EC
Close interactions between the EGFP-labeled hMSC and EC were observed only sporadically in the cocultures propagated in medium M2 for 21 days. A large majority of EC developed separate colonies (Fig. 6). In addition, we observed large relatively loose colonies of EC associated with few hMSC (Fig. 6, colony1), small dense EC clusters amidst sparsely distributed hMSC (Fig. 6, colony 2) as well as compact EC colonies of EC covered with few hMSC (Fig. 6, colony 3) were discerned. The scarcity of direct contacts between the two cell types was obvious also after immunofluorescence staining of CD31-positive cells (Fig. 7). Merging of cells of different type was sporadic. However, occasionally we observed both EC cells enclosed in a cluster of hMSC (Fig. 7E) and isolated hMSC enclosed in a cluster of EC (Fig. 7D).
By time-lapse imaging, we could observe both hMSC and EC actively migrating to join the cells of the same type to finally form clusters (Fig. 8). We could observe that, after adhesion followed by a period of active orientation of several hours, the cells started to migrate to join the cells of the same type (Fig. 8).
DISCUSSION
Aiming at the establishment of cocultures of hMSC and EC for purposes of tissue engineering of bone, we systematically examined the growth characteristics of hMSC in medium supporting the growth of both hMSC and EC. In order to assess possible donor dependent variations of hMSC characteristics and functioning we analysed hMSC from 3 independent donors anticipating that hMSC isolated from the same donor as EC would react differently in cell culture. The cultures of hMSC proliferated continuously reaching the population doubling of up to 10 in the course of the observation period of 21 days. The early development similar to that reported for the hMSC of average young donors having the capacity to undergo 24-40 population doublings [35]. However, in our experiments, the propagation rate of hMSC slowed down in the course of 21 days in the coculture medium M2 (Fig. 2). Even thought the observation period did not exceed 21 days, we could observe extensive differences in the proliferation rate of hMSC isolated from different donors and this confirms previous reports [35].
Independently of the differences in their growth potential, the hMSC from all three donors responded similarly to the induction toward osteogenic, chondrogenic, and adipogenic lineages. As demonstrated by both immunofluorescence and semi-quantitative RT-PCR, only cells propagated M1 presented the full differentiation potential of hMSC. The capacity to differentiate along either chondrogenic or osteogenic lineages was reduced in hMSC propagated in M2 and M3. In contrast, the multipotency of hMSC propagated in Ham’s F-12 medium was reported to be retained up to 23 population doublings [36]. In addition, hMSC preserved their typical morphology for 38 population doublings in medium D-MEM [37]. Interestingly, osteogenic differentiation of hMSC was reported to be preserved despite apparent cell senescence and slowdown in the proliferation rate [36,37]. However a progressive loss of adipogenic and chondrogenic potential in the course of propagation was observed in hMSC-derived clones [38].
We could not detect any expression of either CD31 or CD144 on the surface of hMSC from 3 different donors cultured in M2 containing endothelial cell growth supplements for 21 days. Bone marrow-derived hMSC we used may differ in this respect from the human umbilical blood cord–derived hMSC reported to differentiate along an endothelial lineage in vitro and to acquire the capacity to express a variety of endothelial lineage surface markers after an incubation of 3 weeks in the presence of vascular endothelial growth factor [21], EGF, and hydrocortisone [39]. In addition, hMSC were demonstrated to undergo a medium-dependent differentiation toward EC within 5 days acquiring the capacity to express the endothelial VEGF- receptor Flk2 in coculture systems [40]. Differentiation of hMSC into EC and capacity to express CD31, vWF, and CD144 was also reported to be induced by exposure of hMSC to horse serum and ß-mercaptoethanol for 2 weeks [41]. The importance of the coculture medium components for the preservation of phenotypic characteristics of hMSC emerges also from the report on stimulation of the migration and invasive properties of hMSC by VEGF-A [42].
In our experiments, both hMSC and EC formed distinct clusters and the contacts between two cell types were only sporadic. We observed migration of cells joining the cells of their own type (Fig. 7). A similar re-distribution of the cells was previously reported in spheroids of EC and human osteoblasts [43]. In contrast, in cell-overloaded cocultures of hMSC and EC, a tight integration of hMSC into the endothelial monolayer involving the abolishment of the tight junctions was observed after only 2 hours of coculture [44]. A firm adhesion of hMSC to TNF-α prestimulated EC was also observed both in flow chamber and confirmed in vivo in a mouse model [45]. Interestingly, hMSC, when co-implanted with endothelial cells in a fibronectin-containing collagen gel, were recently found to assume the role of perivascular cells and this way contribute to the vasculogenesis process [46].
Our systematic study on the development of hMSC and EC in coculture system shows an obvious necessity for further optimisation. Even though the coculture medium supported the proliferation of both hMSC and EC, and, in addition, hMSC preserved their capacity to differentiate along both osteogenic and adipogenic lineages, the donor-related differences in the proliferation rate of hMSC as well as their constrained to differentiate along chondrogenic lineage in this medium should be taken into account.
The scarcity of close interactions of hMSC and EC in cocultures suggests that indirect growth factor- mediated rather than direct interactions may be involved in the reciprocal stimulation of both cell types.
CONCLUSION
Aiming at the generation of vascularized bone replacement implants, we examined the interactions of hMSC and EC in cocultures. To evaluate the host-dependent variations, all experiment were performed with hMSC from 3 independent donors. As endothelial cell model, we used the cell line AS-M.5.
In order to find an appropriate culture medium supporting an optimal growth of both cell types, we analyzed the capacity of hMSC to proliferate and differentiate in 3 different growth media. These included media designed for either hMSC or for EC, and a medium composed of equal parts of both. The hMSC cultures of the three donors tested showed considerable differences in growth rate yet responded similarly to the three growth media tested. In addition, all hMSC responded similarly to the induction toward osteogenic, chondrogenic, and adipogenic lineages when propagated in the three growth media for 21 days and subsequently subjected to treatments known to induce osteogenic, chondrogenic, and adipogenic differentiation.
The full differentiation capacity of hMSC was preserved solely in the hMSC medium. However, since all hMSC tested showed the capacity of osteogenic differentiation in the mixed medium, we used this medium in cocultures of hMSC and EC.
In order to identify and to follow up the cells in cocultures, we labelled EC with enhanced green fluorescent protein (EGFP). By time-lapse imaging over 12 h, we observed both hMSC and EC actively migrating to join the cells of their own type to form separate hMSC and EC clusters within 3 weeks. The scarcity of close interaction between hMSC and EC in cocultures suggest growth factor mediated cell interactions and imply the necessity of a careful optimalization of coculture conditions.
ACKNOWLEDGEMENTS
The study was supported by Stiftung Synos and by the AO Research Fund (grant: AO-05-D83) of the AO Foundation. We thank Martina Burggraf for the excellent technical assistance.

